

A variational algorithm to detect the clonal copy number substructure of tumors from scRNA-seq data
- PMID: 36841879
- PMCID: PMC9968345
- DOI: 10.1038/s41467-023-36790-9
A variational algorithm to detect the clonal copy number substructure of tumors from scRNA-seq data
Abstract
Single-cell RNA sequencing is the reference technology to characterize the composition of the tumor microenvironment and to study tumor heterogeneity at high resolution. Here we report Single CEll Variational ANeuploidy analysis (SCEVAN), a fast variational algorithm for the deconvolution of the clonal substructure of tumors from single-cell RNA-seq data. It uses a multichannel segmentation algorithm exploiting the assumption that all the cells in a given copy number clone share the same breakpoints. Thus, the smoothed expression profile of every individual cell constitutes part of the evidence of the copy number profile in each subclone. SCEVAN can automatically and accurately discriminate between malignant and non-malignant cells, resulting in a practical framework to analyze tumors and their microenvironment. We apply SCEVAN to datasets encompassing 106 samples and 93,322 cells from different tumor types and technologies. We demonstrate its application to characterize the intratumor heterogeneity and geographic evolution of malignant brain tumors.
© 2023. The Author(s).
Conflict of interest statement
A.I. received sponsored research funding from AstraZeneca and Taiho Pharmaceutical and has served as a paid consultant/advisor to AIMEDBIO. The remaining authors declare no competing interests.
Figures


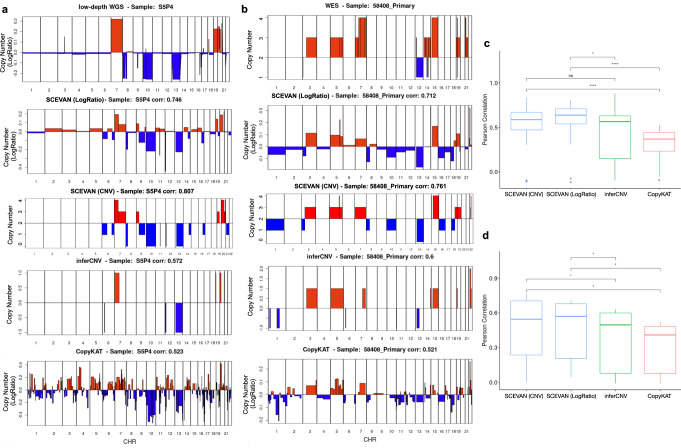

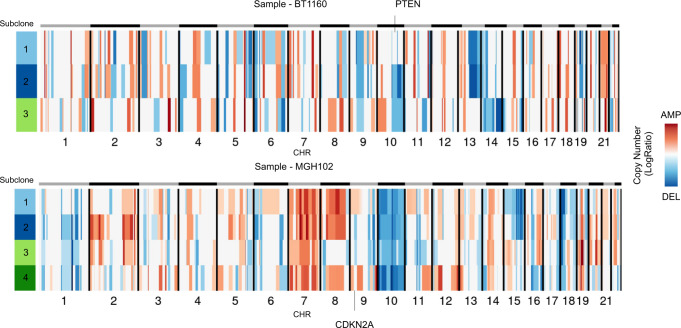
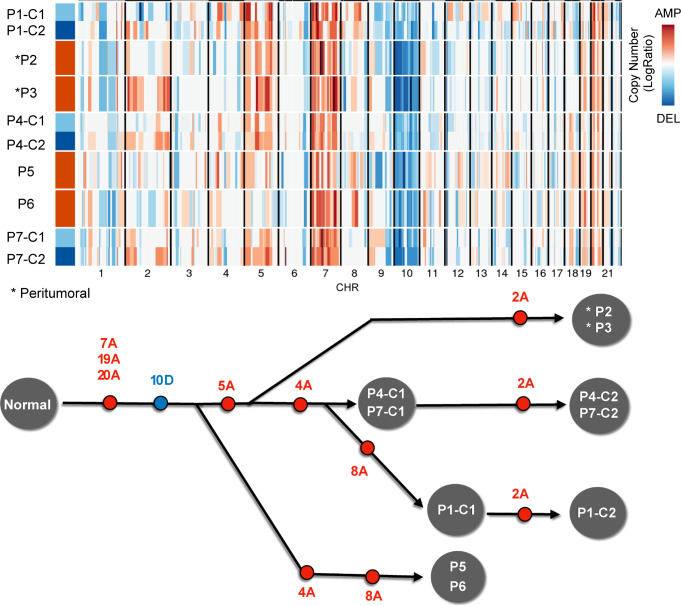

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
