

CRISPR/Cas9-based Engineering of Immunoglobulin Loci in Hybridoma Cells
- PMID: 36845533
- PMCID: PMC9947551
- DOI: 10.21769/BioProtoc.4613
CRISPR/Cas9-based Engineering of Immunoglobulin Loci in Hybridoma Cells
Abstract
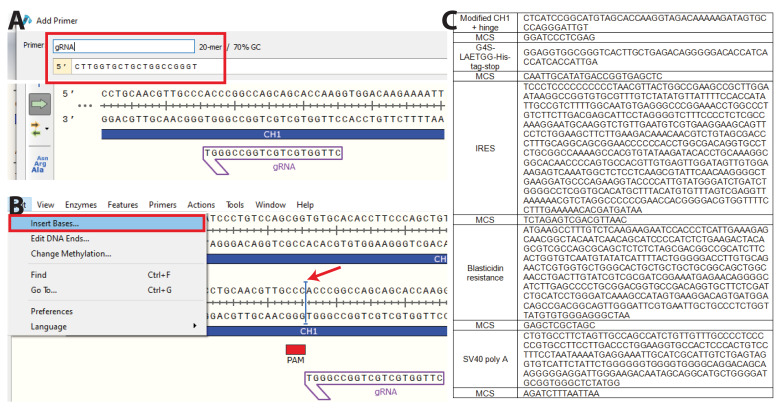
Development of the hybridoma technology by Köhler and Milstein (1975) has revolutionized the immunological field by enabling routine use of monoclonal antibodies (mAbs) in research and development efforts, resulting in their successful application in the clinic today. While recombinant good manufacturing practices production technologies are required to produce clinical grade mAbs, academic laboratories and biotechnology companies still rely on the original hybridoma lines to stably and effortlessly produce high antibody yields at a modest price. In our own work, we were confronted with a major issue when using hybridoma-derived mAbs: there was no control over the antibody format that was produced, a flexibility that recombinant production does allow. We set out to remove this hurdle by genetically engineering antibodies directly in the immunoglobulin (Ig) locus of hybridoma cells. We used clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) and homology-directed repair (HDR) to modify antibody's format [mAb or antigen-binding fragment (Fab')] and isotype. This protocol describes a straightforward approach, with little hands-on time, leading to stable cell lines secreting high levels of engineered antibodies. Parental hybridoma cells are maintained in culture, transfected with a guide RNA (gRNA) targeting the site of interest in the Ig locus and an HDR template to knock in the desired insert and an antibiotic resistance gene. By applying antibiotic pressure, resistant clones are expanded and characterized at the genetic and protein level for their ability to produce modified mAbs instead of the parental protein. Finally, the modified antibody is characterized in functional assays. To demonstrate the versatility of our strategy, we illustrate this protocol with examples where we have (i) exchanged the constant heavy region of the antibody, creating chimeric mAb of a novel isotype, (ii) truncated the antibody to create an antigenic peptide-fused Fab' fragment to produce a dendritic cell-targeted vaccine, and (iii) modified both the constant heavy (CH)1 domain of the heavy chain (HC) and the constant kappa (Cκ) light chain (LC) to introduce site-selective modification tags for further derivatization of the purified protein. Only standard laboratory equipment is required, which facilitates its application across various labs. We hope that this protocol will further disseminate our technology and help other researchers. Graphical abstract.
Keywords: Antibody engineering; CRISPR/Cas9; Hybridoma; Immunoglobulin; Immunology; Immunotherapy.
Copyright © 2023 The Authors; This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
Conflict of interest statement
Competing interestsThe authors declare no competing interest.
Figures







References
-
- Adhikari P., Zacharias N., Ohri R. and Sadowsky J.(2020). Site-Specific Conjugation to Cys-Engineered THIOMABTM Antibodies. In: Tumey, L.(Ed.). Antibody-Drug Conjugates. Methods in Molecular Biology. pp. 51-69. - PubMed
-
- Evers M., Stip M., Keller K., Willemen H., Nederend M., Jansen M., Chan C., Budding K., Nierkens S., Valerius T., et al. .(2021). Anti-GD2 IgA kills tumors by neutrophils without antibody-associated pain in the preclinical treatment of high-risk neuroblastoma. J Immunother Cancer 9(10): e003163. - PMC - PubMed
-
- Fennemann F. L., Le Gall C. M., van der Schoot J. M. S., Ramos-Tomillero I., Figdor C. G., Scheeren F. A., and Verdoes M.(2021). Generation of dendritic cell targeted anti-cancer vaccines by CRISPR- Cas9-editing of the hybridoma immunoglobulin locus. In: Molecularly Defined Dendritic Cell-Targeted Vaccines for Cancer Immunotherapy. pp. 42-63.
LinkOut - more resources
Full Text Sources
