

Refining epigenetic prediction of chronological and biological age
- PMID: 36855161
- PMCID: PMC9976489
- DOI: 10.1186/s13073-023-01161-y
Refining epigenetic prediction of chronological and biological age
Abstract
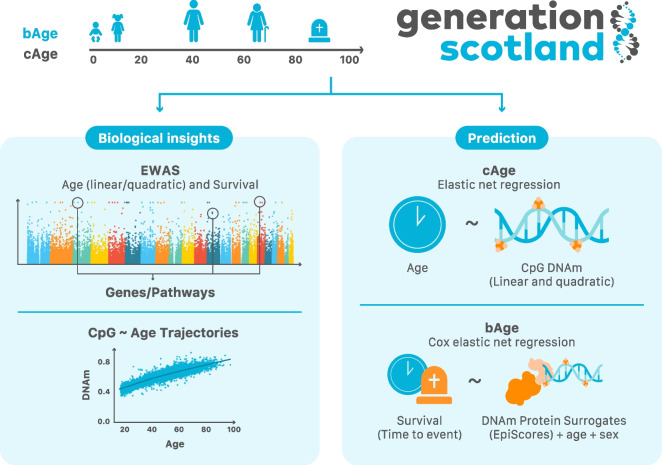
Background: Epigenetic clocks can track both chronological age (cAge) and biological age (bAge). The latter is typically defined by physiological biomarkers and risk of adverse health outcomes, including all-cause mortality. As cohort sample sizes increase, estimates of cAge and bAge become more precise. Here, we aim to develop accurate epigenetic predictors of cAge and bAge, whilst improving our understanding of their epigenomic architecture.
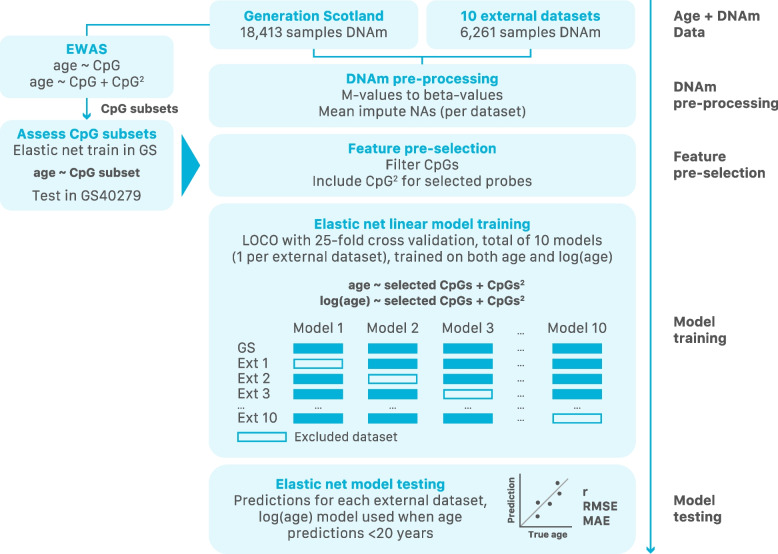
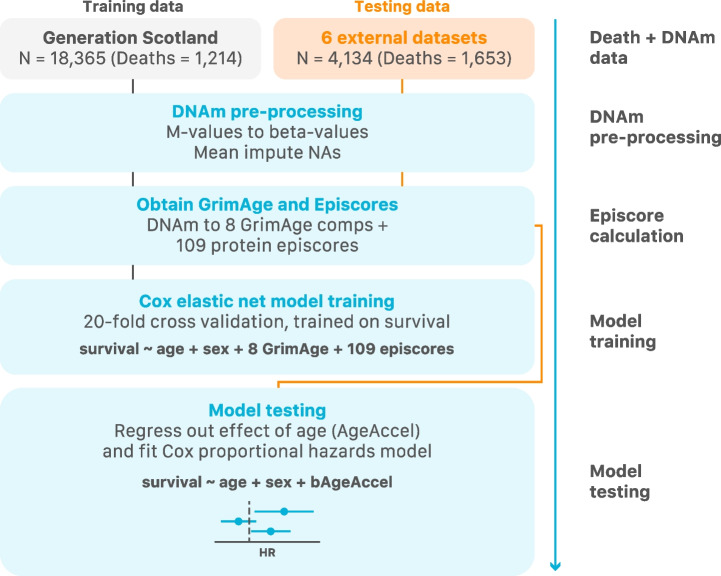
Methods: First, we perform large-scale (N = 18,413) epigenome-wide association studies (EWAS) of chronological age and all-cause mortality. Next, to create a cAge predictor, we use methylation data from 24,674 participants from the Generation Scotland study, the Lothian Birth Cohorts (LBC) of 1921 and 1936, and 8 other cohorts with publicly available data. In addition, we train a predictor of time to all-cause mortality as a proxy for bAge using the Generation Scotland cohort (1214 observed deaths). For this purpose, we use epigenetic surrogates (EpiScores) for 109 plasma proteins and the 8 component parts of GrimAge, one of the current best epigenetic predictors of survival. We test this bAge predictor in four external cohorts (LBC1921, LBC1936, the Framingham Heart Study and the Women's Health Initiative study).
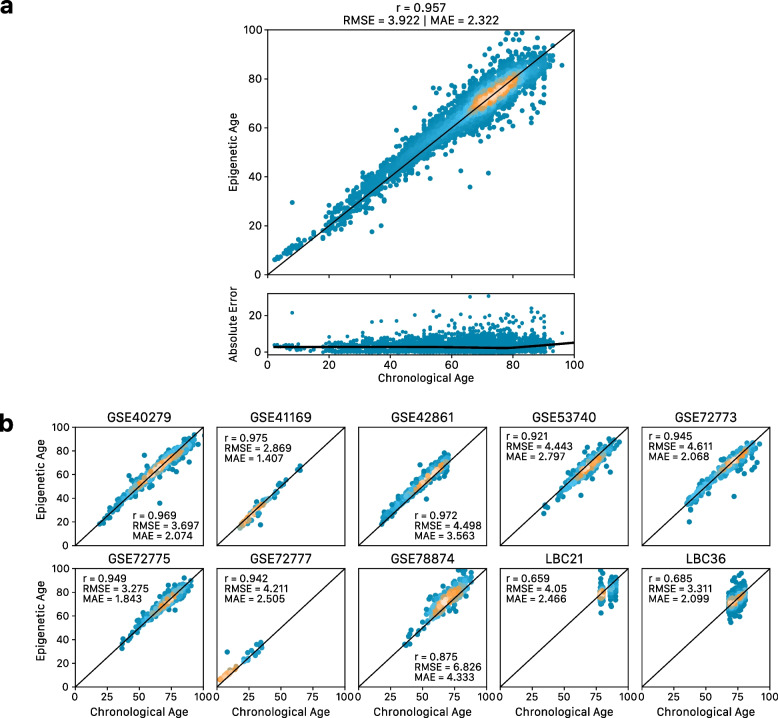
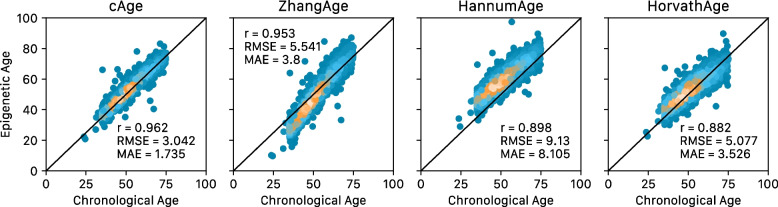
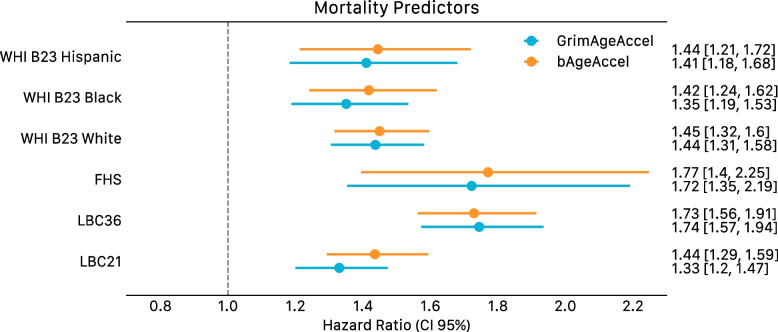
Results: Through the inclusion of linear and non-linear age-CpG associations from the EWAS, feature pre-selection in advance of elastic net regression, and a leave-one-cohort-out (LOCO) cross-validation framework, we obtain cAge prediction with a median absolute error equal to 2.3 years. Our bAge predictor was found to slightly outperform GrimAge in terms of the strength of its association to survival (HRGrimAge = 1.47 [1.40, 1.54] with p = 1.08 × 10-52, and HRbAge = 1.52 [1.44, 1.59] with p = 2.20 × 10-60). Finally, we introduce MethylBrowsR, an online tool to visualise epigenome-wide CpG-age associations.
Conclusions: The integration of multiple large datasets, EpiScores, non-linear DNAm effects, and new approaches to feature selection has facilitated improvements to the blood-based epigenetic prediction of biological and chronological age.
© 2023. The Author(s).
Conflict of interest statement
R.E.M has received a speaker fee from Illumina and is an advisor to the Epigenetic Clock Development Foundation. R.F.H. has received consultant fees from Illumina. R.E.M, R.F.H., and D.A.G. have received consultant fees from Optima partners. A.M.M has previously received speaker fees from Janssen and Illumina and research funding from The Sackler Trust. M.R.R. receives research funding from Boehringer Ingelheim. All other authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
