

Aggressive Pituitary Tumors and Pituitary Carcinomas: From Pathology to Treatment
- PMID: 36856733
- PMCID: PMC10271233
- DOI: 10.1210/clinem/dgad098
Aggressive Pituitary Tumors and Pituitary Carcinomas: From Pathology to Treatment
Erratum in
-
Correction to "Aggressive Pituitary Tumors and Pituitary Carcinomas: From Pathology to Treatment".J Clin Endocrinol Metab. 2023 Sep 18;108(10):e1163. doi: 10.1210/clinem/dgad222. J Clin Endocrinol Metab. 2023. PMID: 37104780 Free PMC article. No abstract available.
Abstract
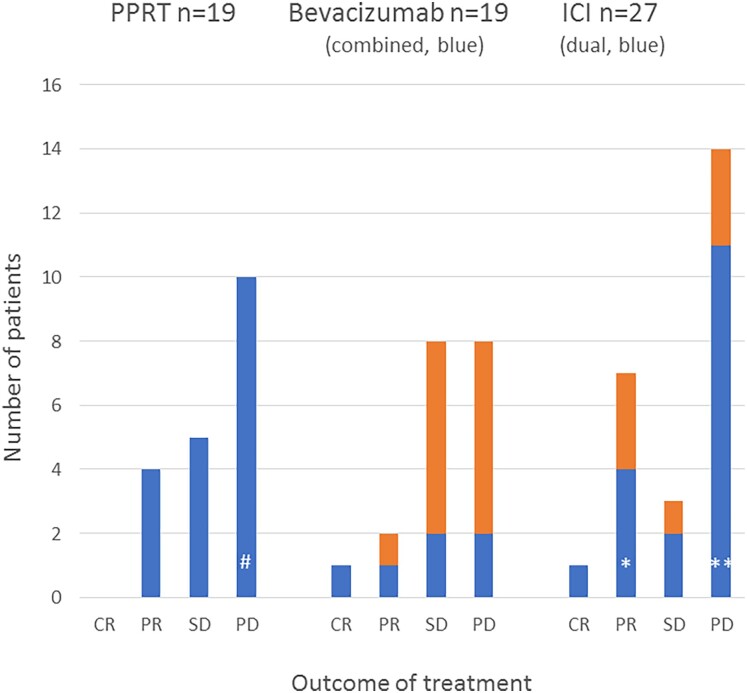
Aggressive pituitary tumors (APTs) and pituitary carcinomas (PCs) are heterogeneous with regard to clinical presentation, proliferative markers, clinical course, and response to therapy. Half of them show an aggressive course only many years after the first apparently benign presentation. APTs and PCs share several properties, but a Ki67 index greater than or equal to 10% and extensive p53 expression are more prevalent in PCs. Mutations in TP53 and ATRX are the most common genetic alterations; their detection might be of value for early identification of aggressiveness. Treatment requires a multimodal approach including surgery, radiotherapy, and drugs. Temozolomide is the recommended first-line chemotherapy, with response rates of about 40%. Immune checkpoint inhibitors have emerged as second-line treatment in PCs, with currently no evidence for a superior effect of dual therapy compared to monotherapy with PD-1 blockers. Bevacizumab has resulted in partial response (PR) in few patients; tyrosine kinase inhibitors and everolimus have generally not been useful. The effect of peptide receptor radionuclide therapy is limited as well. Management of APT/PC is challenging and should be discussed within an expert team with consideration of clinical and pathological findings, age, and general condition of the patient. Considering that APT/PCs are rare, new therapies should preferably be evaluated in shared standardized protocols. Prognostic and predictive markers to guide treatment decisions are needed and are the scope of ongoing research.
Trial registration: ClinicalTrials.gov NCT04244708.
Keywords: ATRX; Ki67- index; PRRT; TP53; bevacizumab; immunotherapy; temozolomide.
© The Author(s) 2023. Published by Oxford University Press on behalf of the Endocrine Society.
Figures








References
-
- Raverot G, Burman P, McCormack A, et al. Clinical practice guidelines for the management of aggressive pituitary tumours and carcinomas. Eur J Endocrinol. 2018;178(1):G1‐G24. - PubMed
-
- WHO Classification of Tumours Editorial Board . Endocrine and Neuroendocrine Tumours [Internet]. International Agency for Research on Cancer; 2022 (WHO Classification of Tumours Series, 5th ed; vol. 8). https://tumourclassification.iarc.who.int/chapters/36
-
- Chanson P, Dormoy A, Dekkers OM. Use of radiotherapy after pituitary surgery for non-functioning pituitary adenomas. Eur J Endocrinol. 2019;181(1):D1‐D13. - PubMed
-
- Tampourlou M, Ntali G, Ahmed S, et al. Outcome of nonfunctioning pituitary adenomas that regrow after primary treatment: a study from two large UK centers. J Clin Endocrinol Metab. 2017;102(6):1889‐1897. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
