

What is cancer metabolism?
- PMID: 36858045
- PMCID: PMC10106389
- DOI: 10.1016/j.cell.2023.01.038
What is cancer metabolism?
Abstract
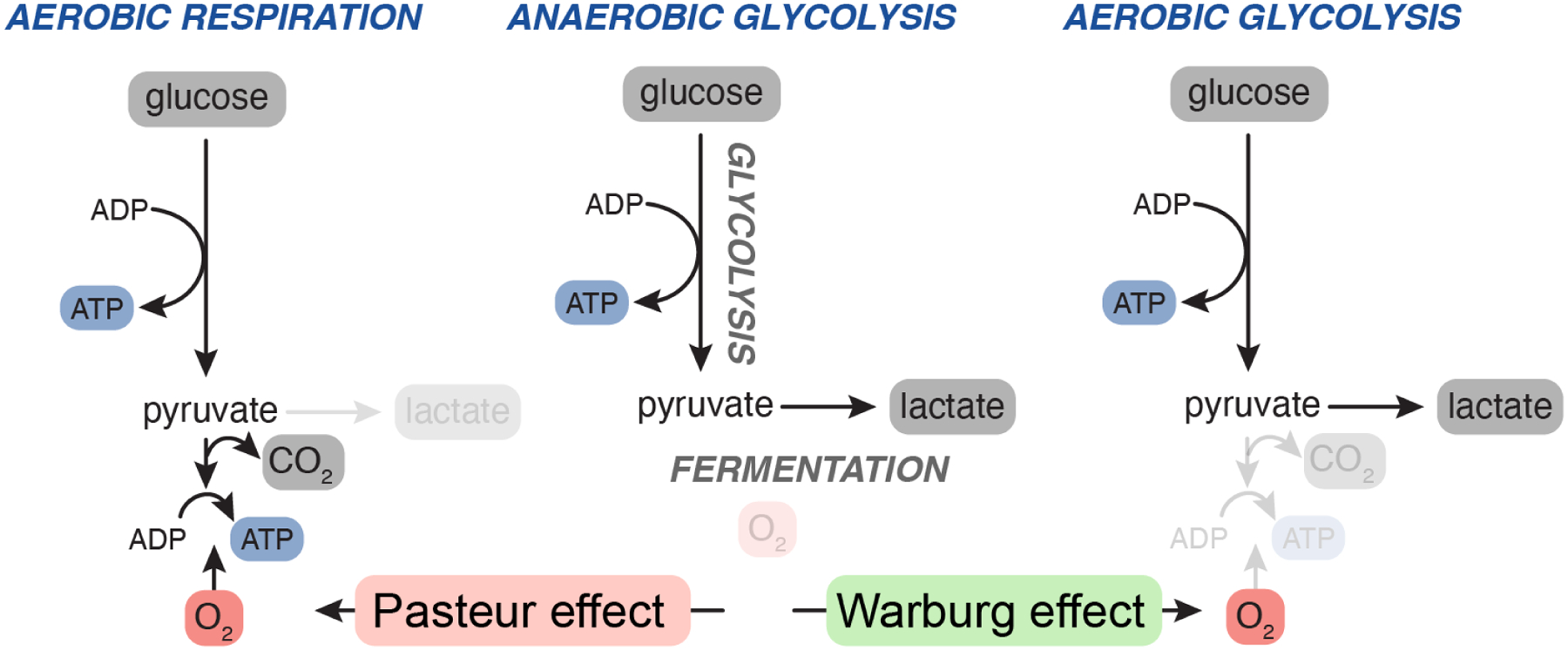
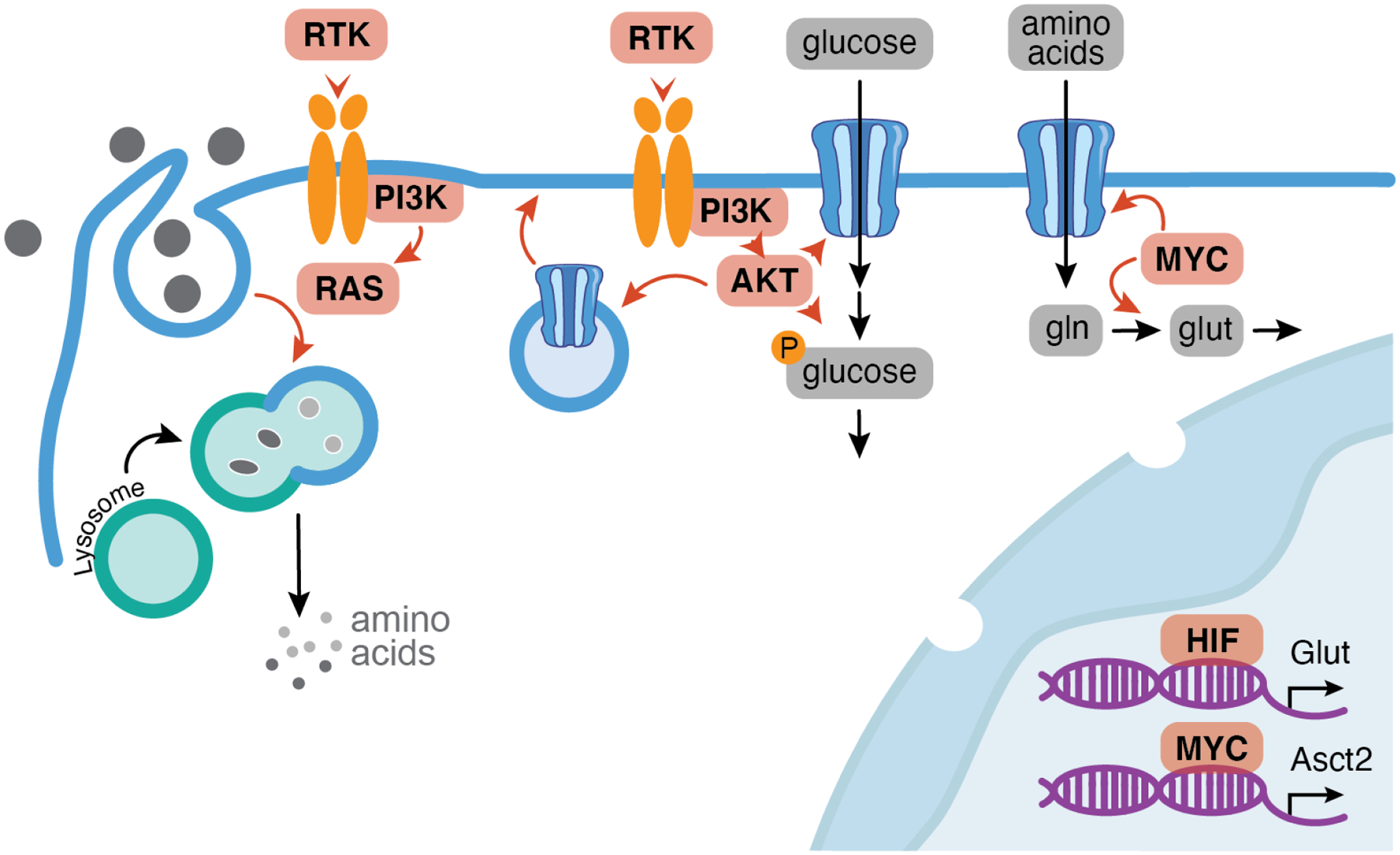
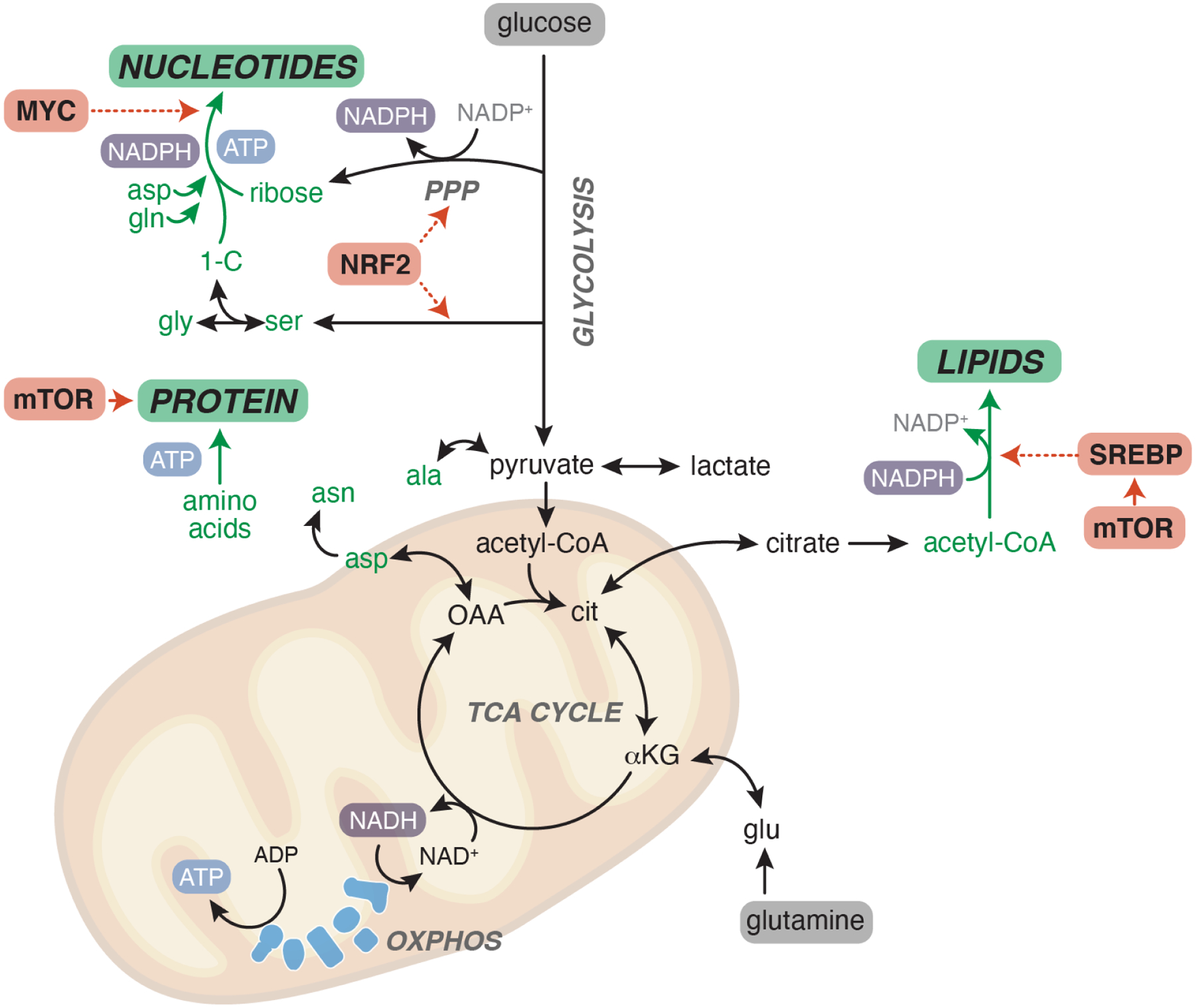
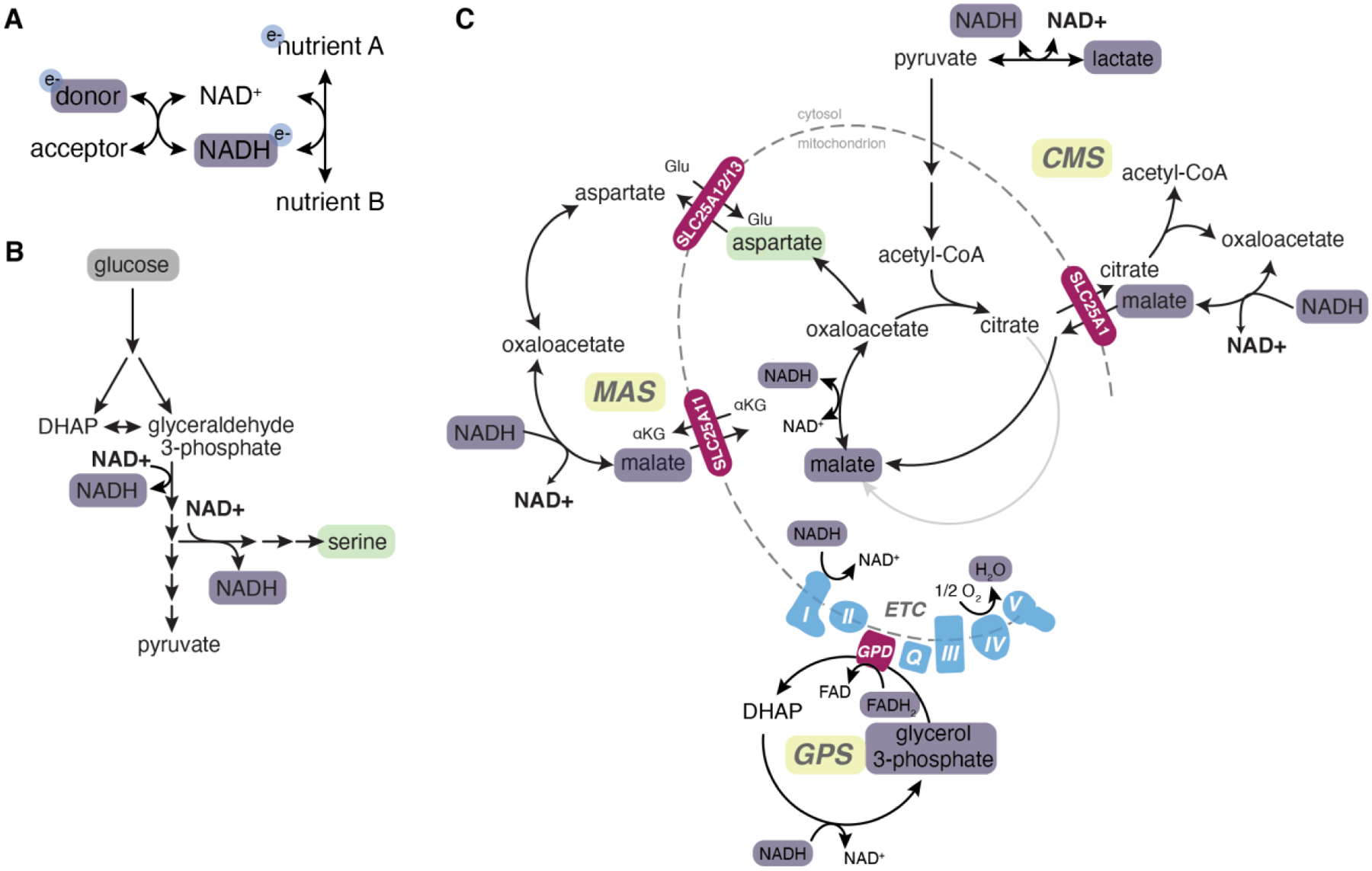
The uptake and metabolism of nutrients support fundamental cellular process from bioenergetics to biomass production and cell fate regulation. While many studies of cell metabolism focus on cancer cells, the principles of metabolism elucidated in cancer cells apply to a wide range of mammalian cells. The goal of this review is to discuss how the field of cancer metabolism provides a framework for revealing principles of cell metabolism and for dissecting the metabolic networks that allow cells to meet their specific demands. Understanding context-specific metabolic preferences and liabilities will unlock new approaches to target cancer cells to improve patient care.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests L.W.S.F. holds a patent and is an author on patent applications related to cellular metabolism and cell fate control.
Figures
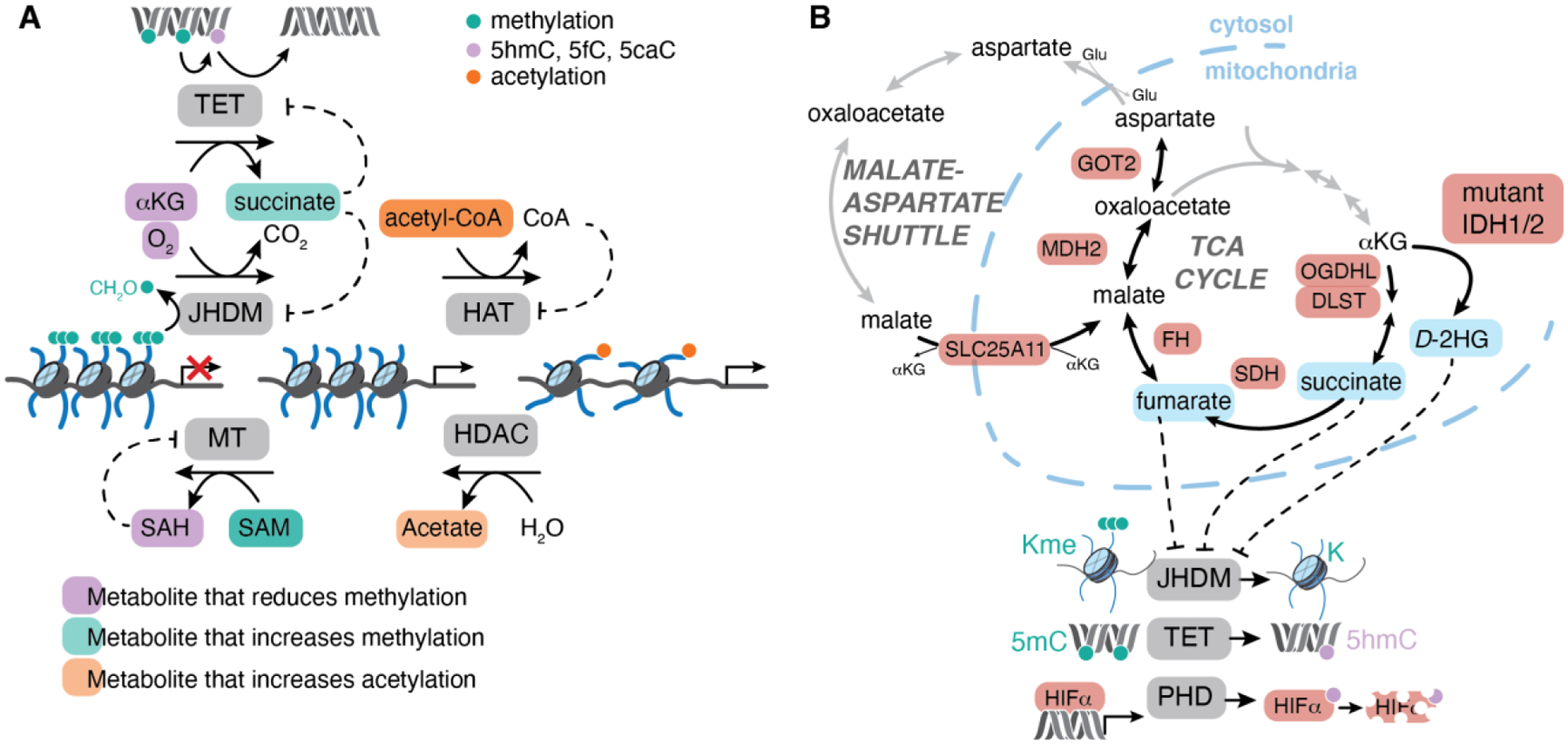
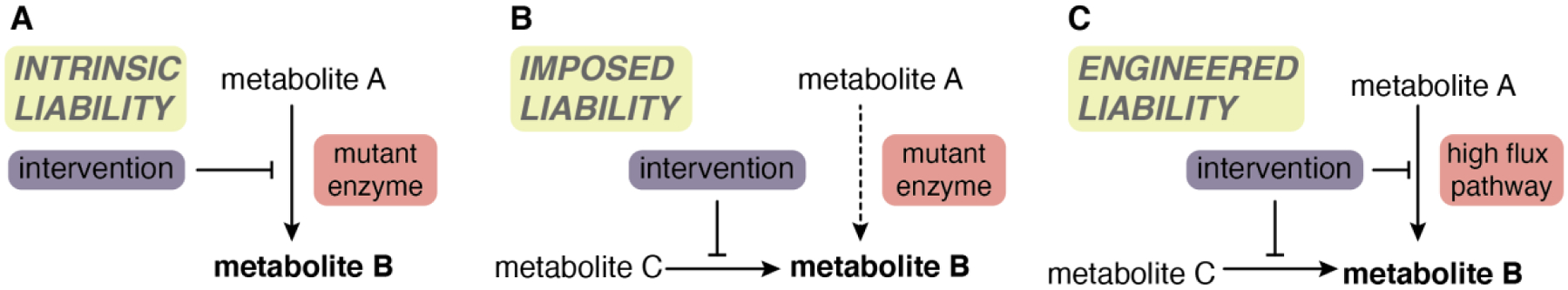
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
