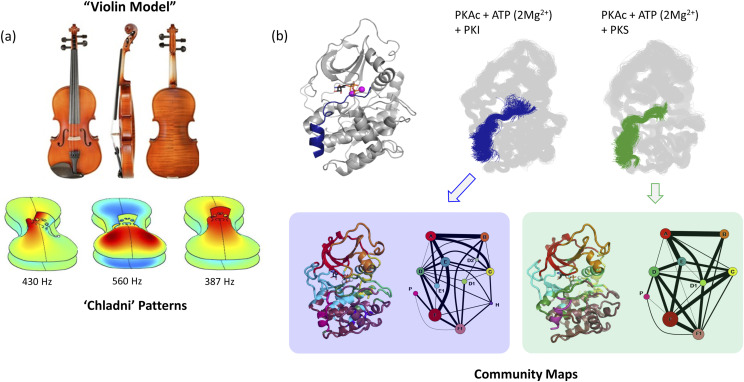
The "violin model": Looking at community networks for dynamic allostery
- PMID: 36859094
- PMCID: PMC9957607
- DOI: 10.1063/5.0138175
The "violin model": Looking at community networks for dynamic allostery
Abstract
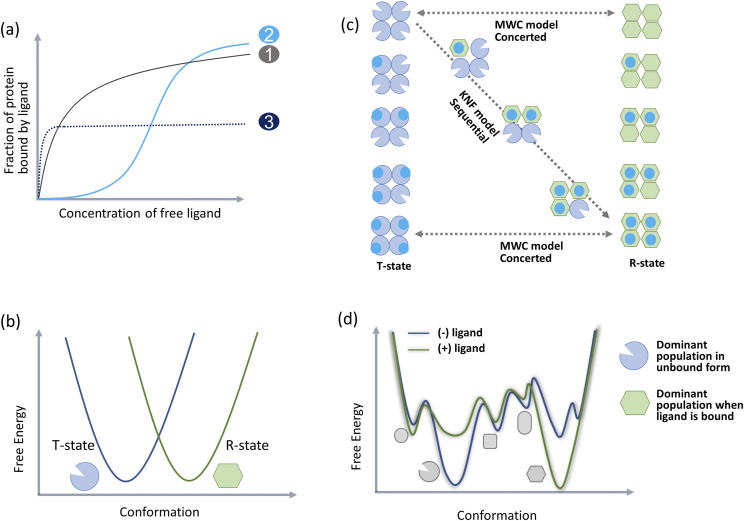
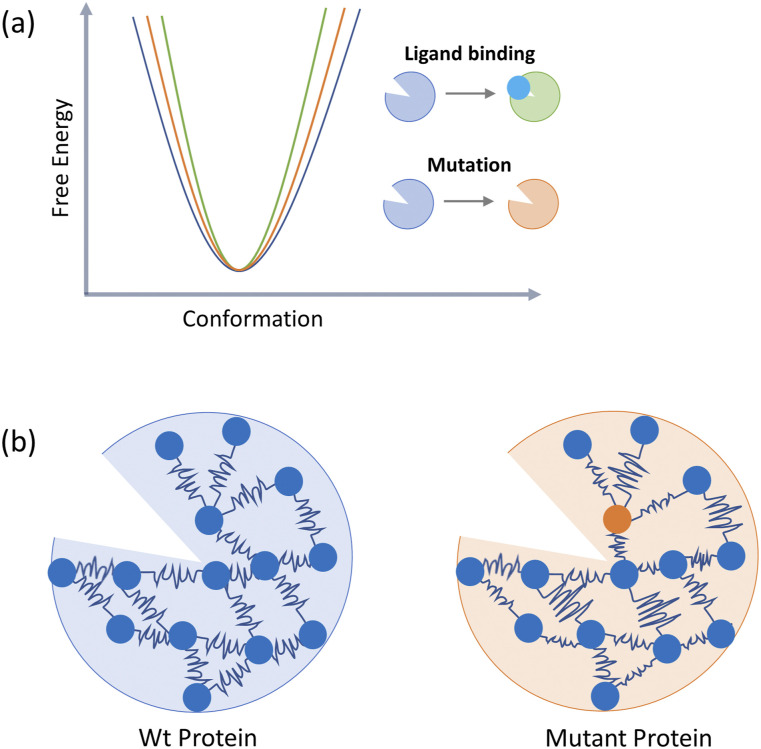
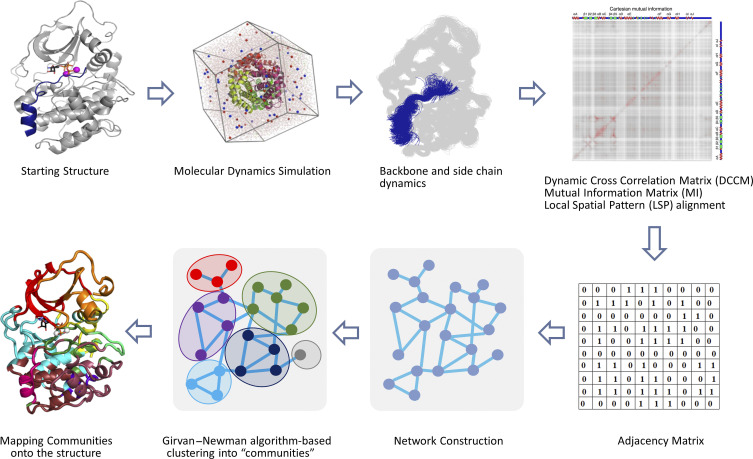
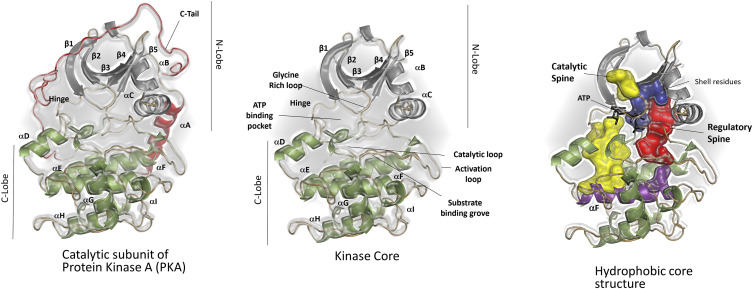
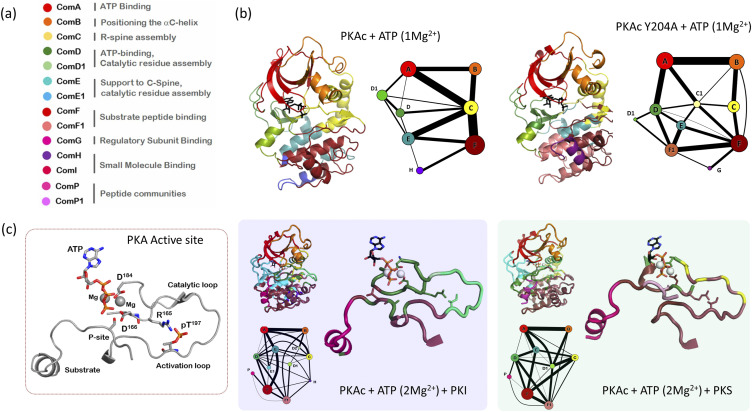
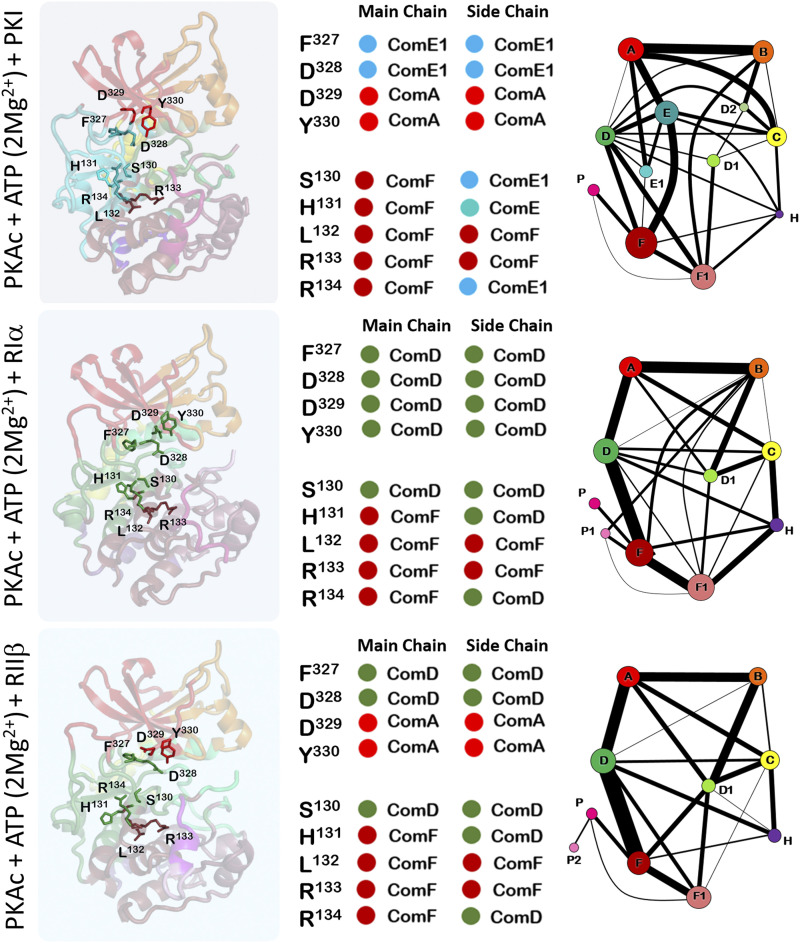
Allosteric regulation of proteins continues to be an engaging research topic for the scientific community. Models describing allosteric communication have evolved from focusing on conformation-based descriptors of protein structural changes to appreciating the role of internal protein dynamics as a mediator of allostery. Here, we explain a "violin model" for allostery as a contemporary method for approaching the Cooper-Dryden model based on redistribution of protein thermal fluctuations. Based on graph theory, the violin model makes use of community network analysis to functionally cluster correlated protein motions obtained from molecular dynamics simulations. This Review provides the theory and workflow of the methodology and explains the application of violin model to unravel the workings of protein kinase A.
Figures