

Computational network analysis of host genetic risk variants of severe COVID-19
- PMID: 36859360
- PMCID: PMC9977643
- DOI: 10.1186/s40246-023-00454-y
Computational network analysis of host genetic risk variants of severe COVID-19
Abstract
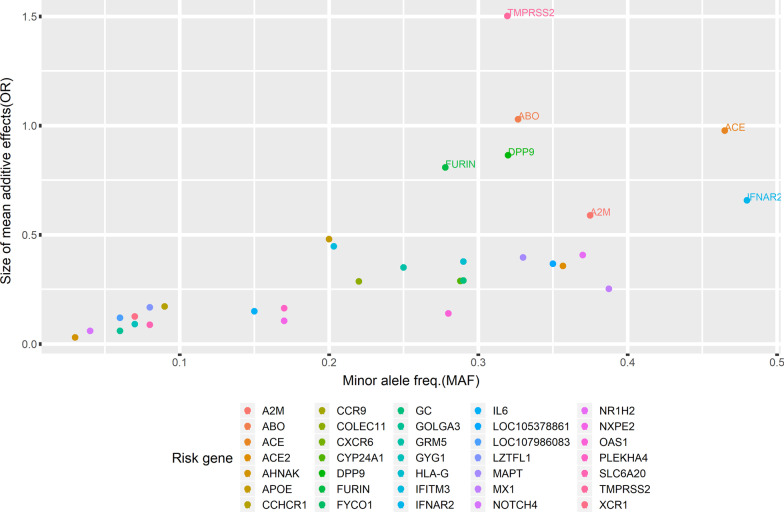
Background: Genome-wide association studies have identified numerous human host genetic risk variants that play a substantial role in the host immune response to SARS-CoV-2. Although these genetic risk variants significantly increase the severity of COVID-19, their influence on body systems is poorly understood. Therefore, we aim to interpret the biological mechanisms and pathways associated with the genetic risk factors and immune responses in severe COVID-19. We perform a deep analysis of previously identified risk variants and infer the hidden interactions between their molecular networks through disease mapping and the similarity of the molecular functions between constructed networks.
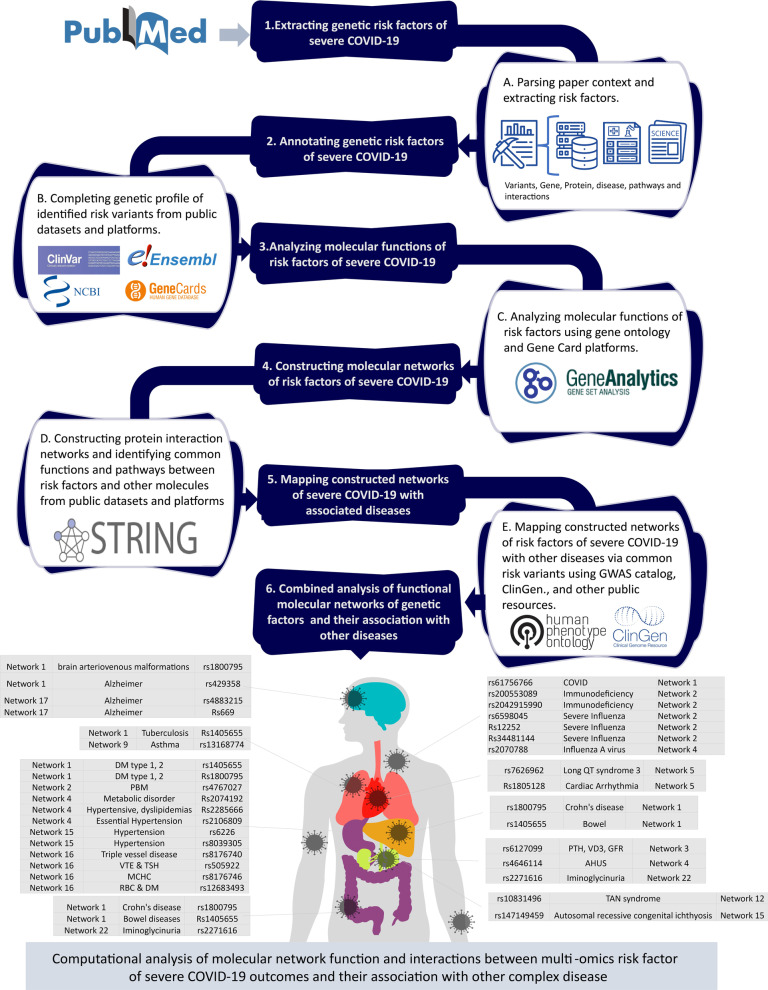
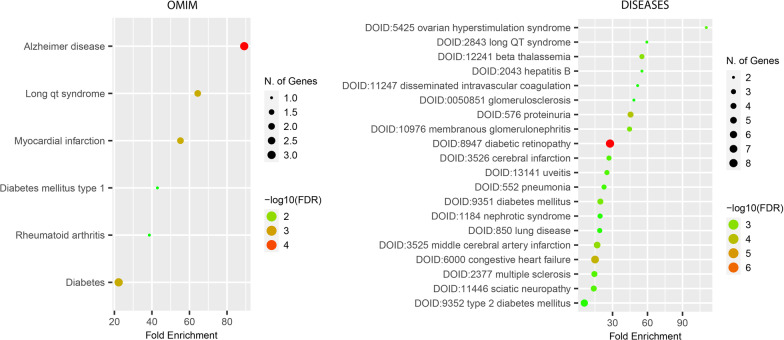
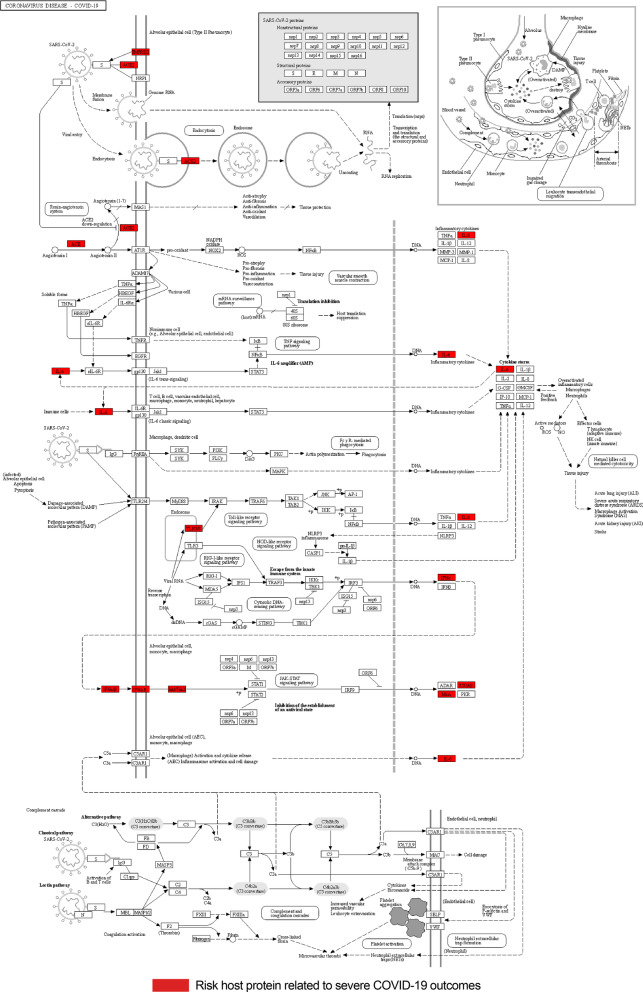
Results: We designed a four-stage computational workflow for systematic genetic analysis of the risk variants. We integrated the molecular profiles of the risk factors with associated diseases, then constructed protein-protein interaction networks. We identified 24 protein-protein interaction networks with 939 interactions derived from 109 filtered risk variants in 60 risk genes and 56 proteins. The majority of molecular functions, interactions and pathways are involved in immune responses; several interactions and pathways are related to the metabolic and cardiovascular systems, which could lead to multi-organ complications and dysfunction.
Conclusions: This study highlights the importance of analyzing molecular interactions and pathways to understand the heterogeneous susceptibility of the host immune response to SARS-CoV-2. We propose new insights into pathogenicity analysis of infections by including genetic risk information as essential factors to predict future complications during and after infection. This approach may assist more precise clinical decisions and accurate treatment plans to reduce COVID-19 complications.
Keywords: Disease mapping; GWAS; Genetic risk factor analysis; Host risk variants; Molecular networks analysis; Severe COVID-19; Statistical analysis.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures








References
-
- Butler-Laporte G, Nakanishi T, Mooser V, Morrison DR, Abdullah T, Adeleye O, Mamlouk N, Kimchi N, Afrasiabi Z, Rezk N, et al. Vitamin D and COVID-19 susceptibility and severity in the COVID-19 host genetics initiative: a mendelian randomization study. PLoS Med. 2021;18(6):1003605. doi: 10.1371/journal.pmed.1003605. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
