

Evolution of the germline mutation rate across vertebrates
- PMID: 36859541
- PMCID: PMC9995274
- DOI: 10.1038/s41586-023-05752-y
Evolution of the germline mutation rate across vertebrates
Abstract
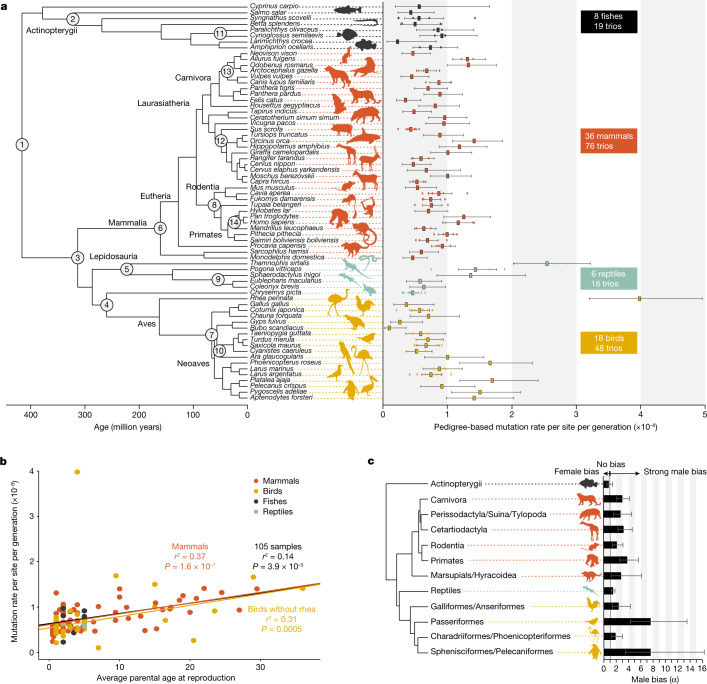
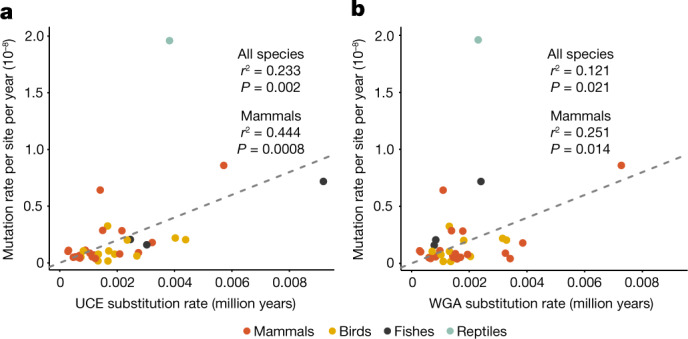
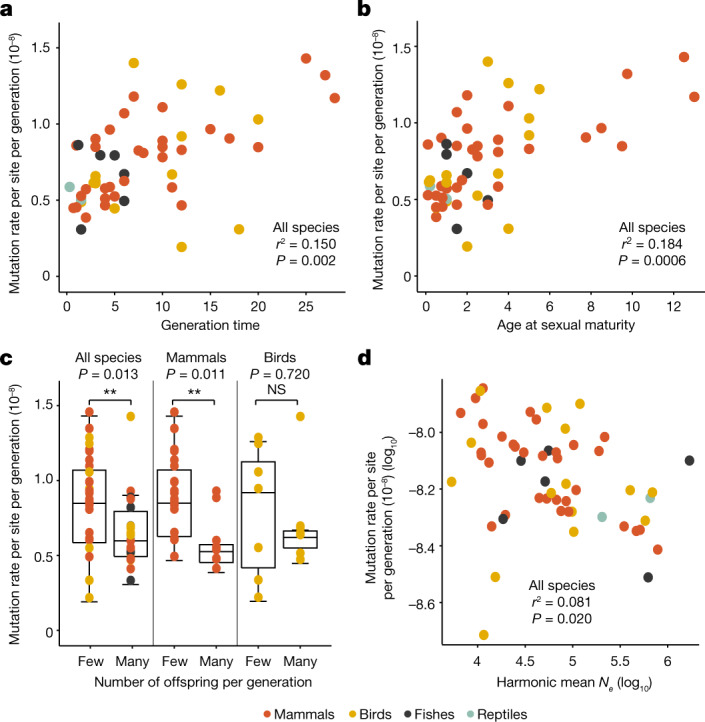
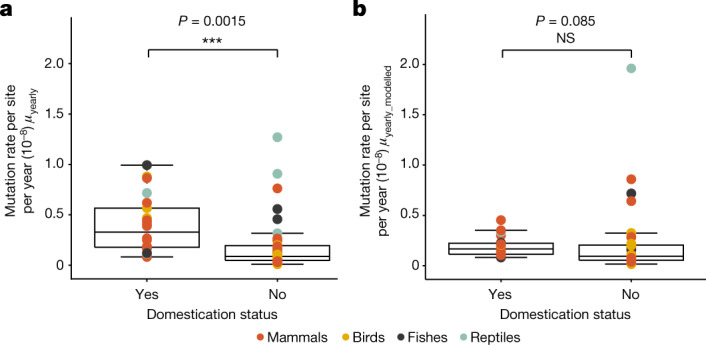
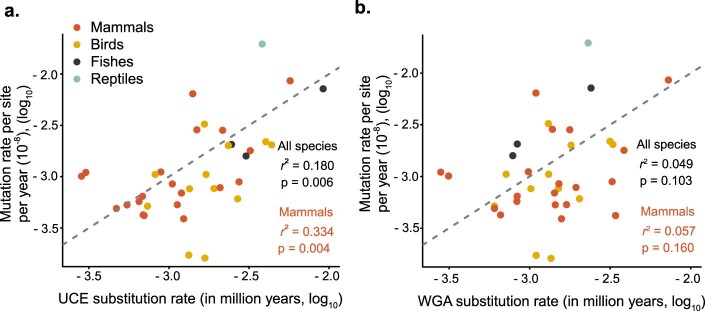
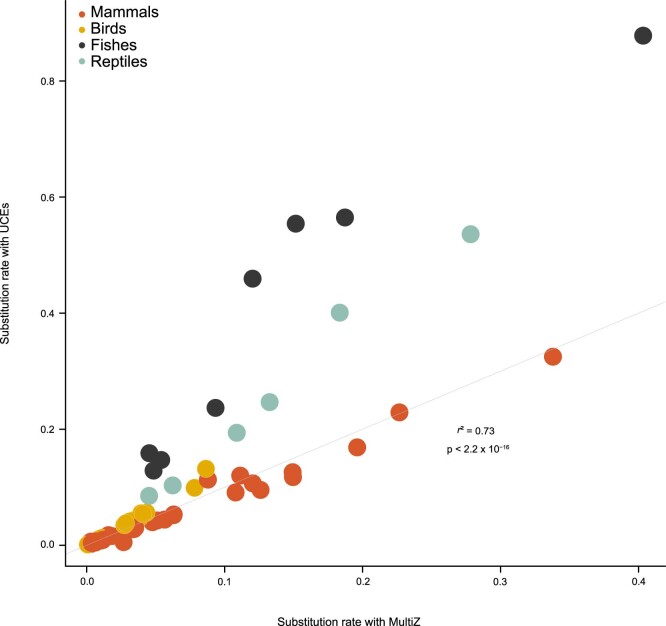
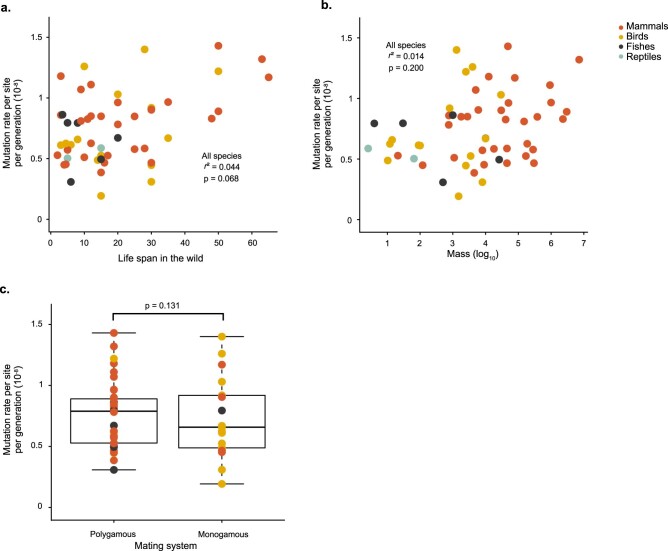
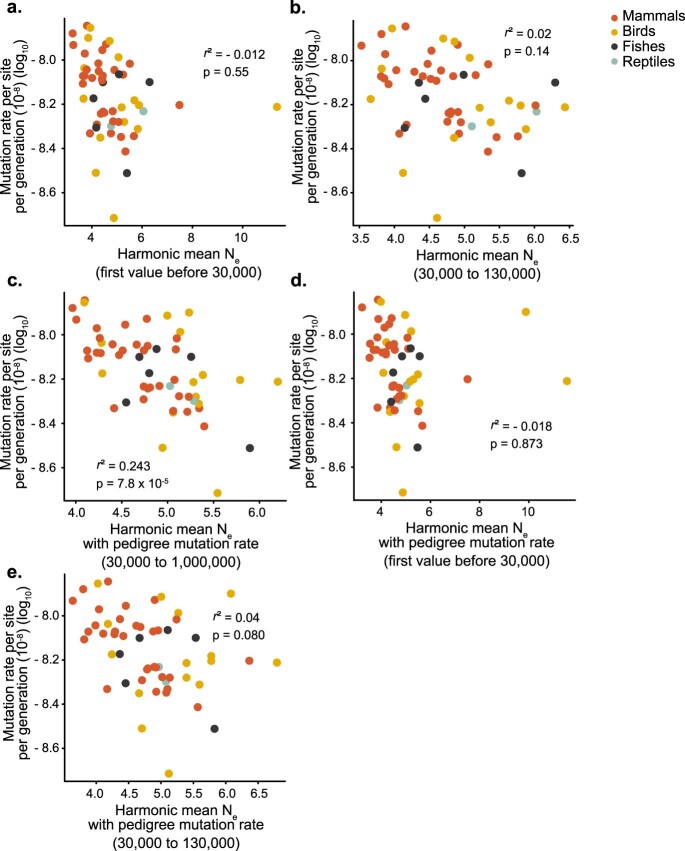
The germline mutation rate determines the pace of genome evolution and is an evolving parameter itself1. However, little is known about what determines its evolution, as most studies of mutation rates have focused on single species with different methodologies2. Here we quantify germline mutation rates across vertebrates by sequencing and comparing the high-coverage genomes of 151 parent-offspring trios from 68 species of mammals, fishes, birds and reptiles. We show that the per-generation mutation rate varies among species by a factor of 40, with mutation rates being higher for males than for females in mammals and birds, but not in reptiles and fishes. The generation time, age at maturity and species-level fecundity are the key life-history traits affecting this variation among species. Furthermore, species with higher long-term effective population sizes tend to have lower mutation rates per generation, providing support for the drift barrier hypothesis3. The exceptionally high yearly mutation rates of domesticated animals, which have been continually selected on fecundity traits including shorter generation times, further support the importance of generation time in the evolution of mutation rates. Overall, our comparative analysis of pedigree-based mutation rates provides ecological insights on the mutation rate evolution in vertebrates.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
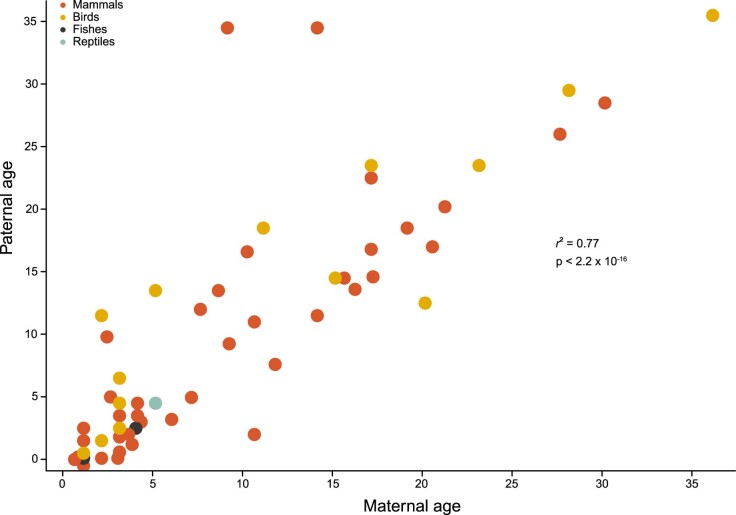
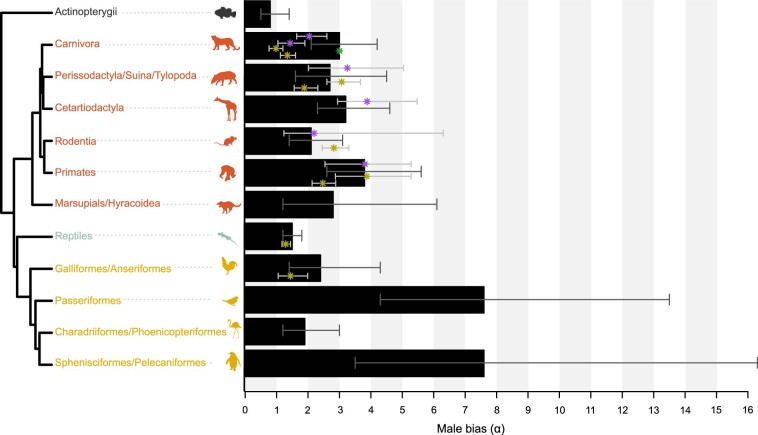
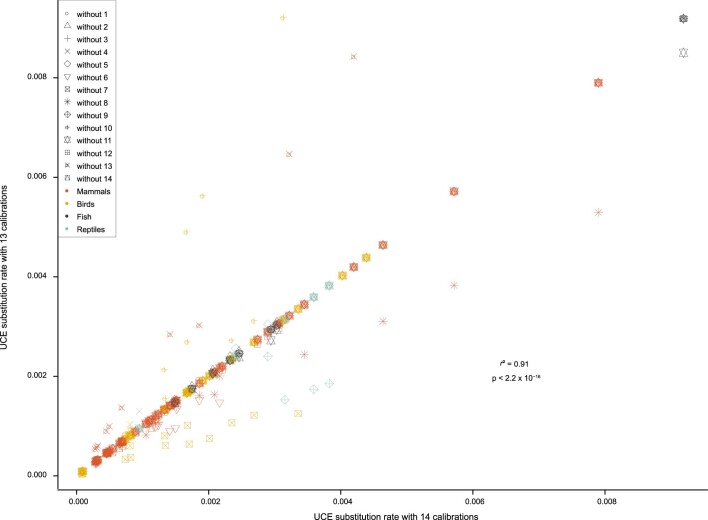
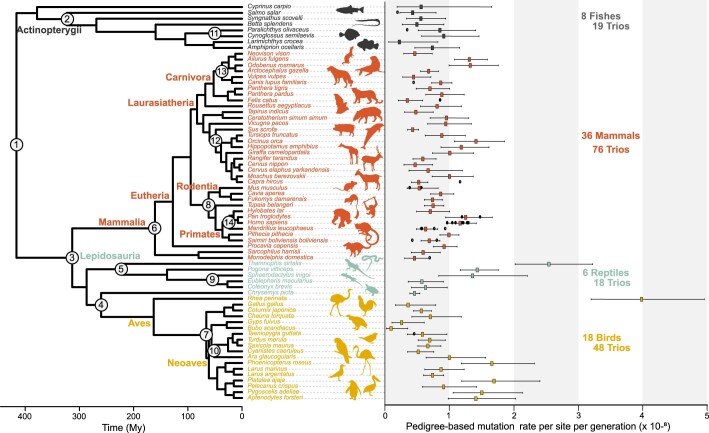
References
-
- Sturtevant AH. Essays on evolution. I. On the effects of selection on mutation rate. Q. Rev. Biol. 1937;12:464–467. doi: 10.1086/394543. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
