

H3K4me3 regulates RNA polymerase II promoter-proximal pause-release
- PMID: 36859550
- PMCID: PMC9995272
- DOI: 10.1038/s41586-023-05780-8
H3K4me3 regulates RNA polymerase II promoter-proximal pause-release
Erratum in
-
Publisher Correction: H3K4me3 regulates RNA polymerase II promoter-proximal pause-release.Nature. 2023 Apr;616(7956):E7. doi: 10.1038/s41586-023-05958-0. Nature. 2023. PMID: 36941349 Free PMC article. No abstract available.
-
Author Correction: H3K4me3 regulates RNA polymerase II promoter-proximal pause-release.Nature. 2023 Nov;623(7987):E8. doi: 10.1038/s41586-023-06778-y. Nature. 2023. PMID: 37907651 Free PMC article. No abstract available.
Abstract
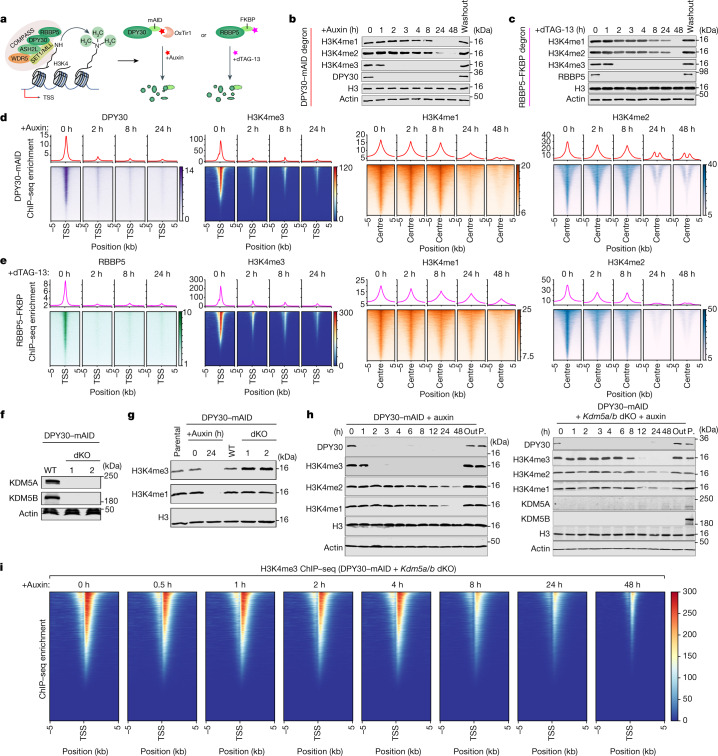
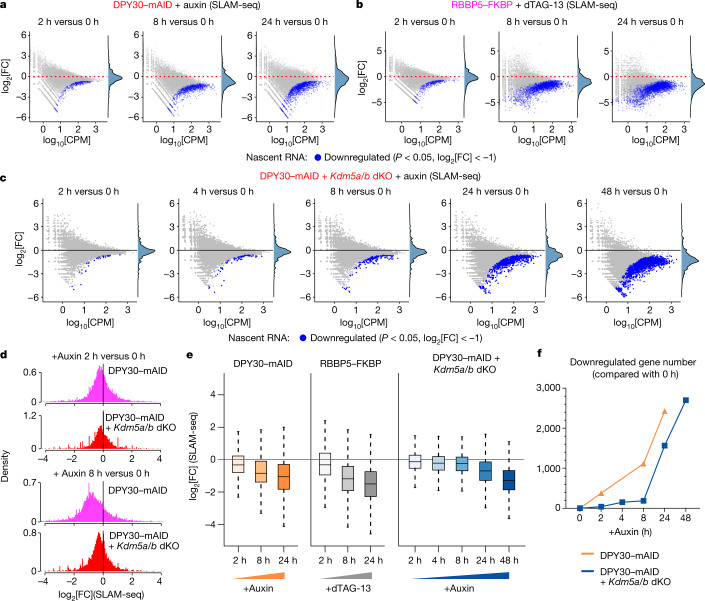
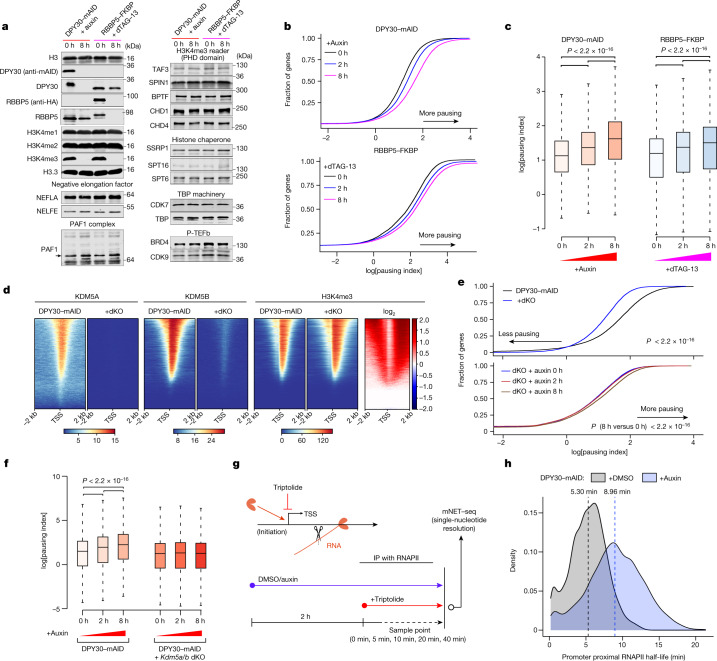
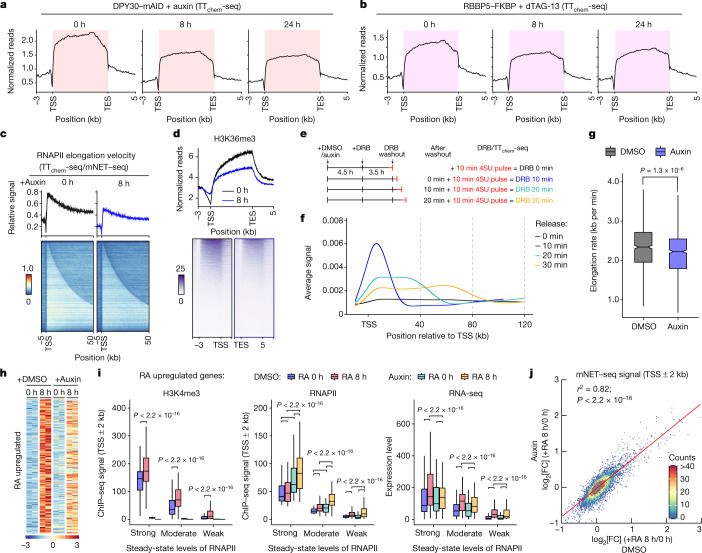
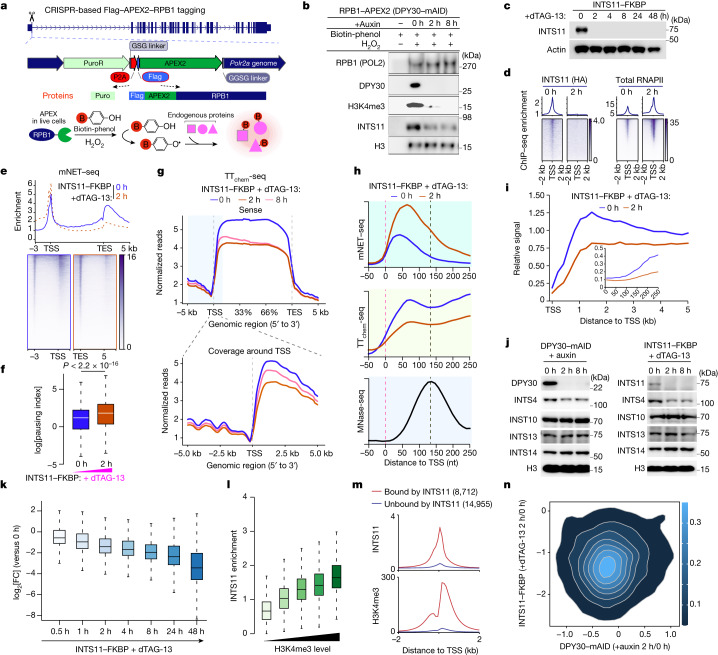
Trimethylation of histone H3 lysine 4 (H3K4me3) is associated with transcriptional start sites and has been proposed to regulate transcription initiation1,2. However, redundant functions of the H3K4 SET1/COMPASS methyltransferase complexes complicate the elucidation of the specific role of H3K4me3 in transcriptional regulation3,4. Here, using mouse embryonic stem cells as a model system, we show that acute ablation of shared subunits of the SET1/COMPASS complexes leads to a complete loss of all H3K4 methylation. Turnover of H3K4me3 occurs more rapidly than that of H3K4me1 and H3K4me2 and is dependent on KDM5 demethylases. Notably, acute loss of H3K4me3 does not have detectable effects on transcriptional initiation but leads to a widespread decrease in transcriptional output, an increase in RNA polymerase II (RNAPII) pausing and slower elongation. We show that H3K4me3 is required for the recruitment of the integrator complex subunit 11 (INTS11), which is essential for the eviction of paused RNAPII and transcriptional elongation. Thus, our study demonstrates a distinct role for H3K4me3 in transcriptional pause-release and elongation rather than transcriptional initiation.
© 2023. The Author(s).
Conflict of interest statement
K.H. is a co-founder of Dania Therapeutics, consultant for Inthera Bioscience and a scientific advisor for MetaboMed and Hannibal Innovation. The other authors declare no competing interests.
Figures











References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
