

Variants in ATP5F1B are associated with dominantly inherited dystonia
- PMID: 36860166
- PMCID: PMC10316767
- DOI: 10.1093/brain/awad068
Variants in ATP5F1B are associated with dominantly inherited dystonia
Abstract
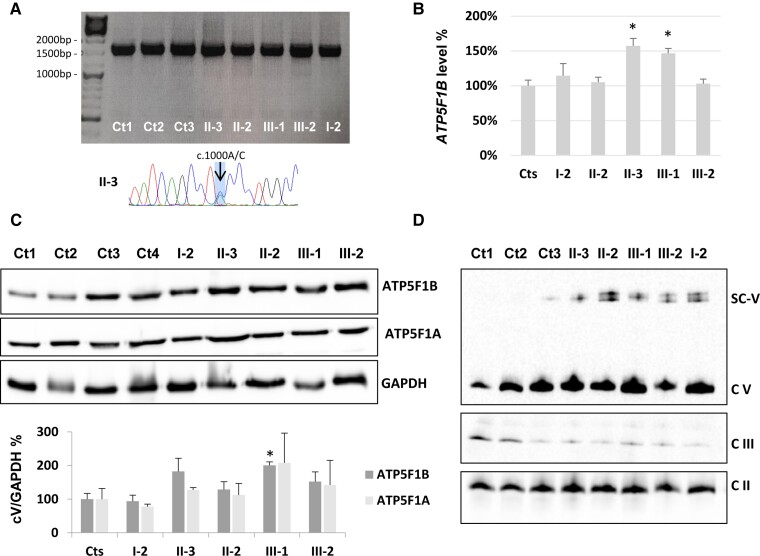
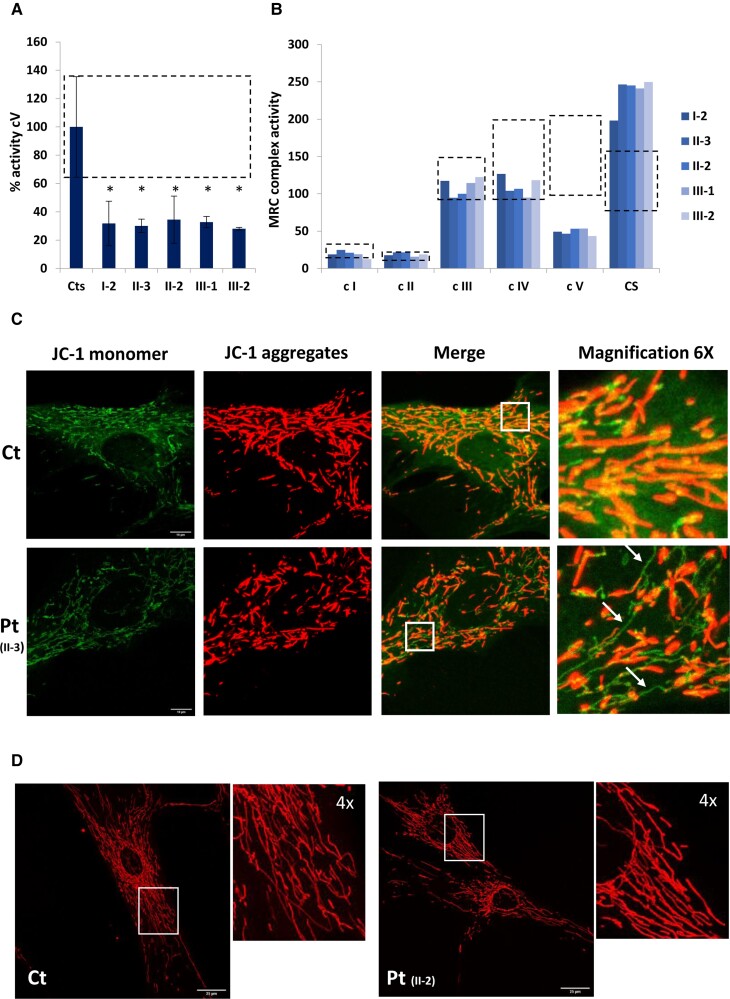
ATP5F1B is a subunit of the mitochondrial ATP synthase or complex V of the mitochondrial respiratory chain. Pathogenic variants in nuclear genes encoding assembly factors or structural subunits are associated with complex V deficiency, typically characterized by autosomal recessive inheritance and multisystem phenotypes. Movement disorders have been described in a subset of cases carrying autosomal dominant variants in structural subunits genes ATP5F1A and ATP5MC3. Here, we report the identification of two different ATP5F1B missense variants (c.1000A>C; p.Thr334Pro and c.1445T>C; p.Val482Ala) segregating with early-onset isolated dystonia in two families, both with autosomal dominant mode of inheritance and incomplete penetrance. Functional studies in mutant fibroblasts revealed no decrease of ATP5F1B protein amount but severe reduction of complex V activity and impaired mitochondrial membrane potential, suggesting a dominant-negative effect. In conclusion, our study describes a new candidate gene associated with isolated dystonia and confirms that heterozygous variants in genes encoding subunits of the mitochondrial ATP synthase may cause autosomal dominant isolated dystonia with incomplete penetrance, likely through a dominant-negative mechanism.
Keywords: ATP5F1B; case report; dystonia; incomplete penetrance; mitochondrial ATP synthase.
© The Author(s) 2023. Published by Oxford University Press on behalf of the Guarantors of Brain.
Conflict of interest statement
The authors report no competing interests.
Figures

References
-
- Schreglmann SR, Riederer F, Galovic M, et al. Movement disorders in genetically confirmed mitochondrial disease and the putative role of the cerebellum: Mitochondrial movement disorders. Mov Disord. 2018;33:146–155. - PubMed
-
- Musumeci O, Oteri R, Toscano A. Spectrum of movement disorders in mitochondrial diseases. J Transl Genet Genomics. 2020;4:221–237.
-
- Fernandez-Vizarra E, Zeviani M. Mitochondrial disorders of the OXPHOS system. FEBS Lett. 2021;595:1062–1106. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
