

CEP162 deficiency causes human retinal degeneration and reveals a dual role in ciliogenesis and neurogenesis
- PMID: 36862503
- PMCID: PMC10104899
- DOI: 10.1172/JCI161156
CEP162 deficiency causes human retinal degeneration and reveals a dual role in ciliogenesis and neurogenesis
Abstract
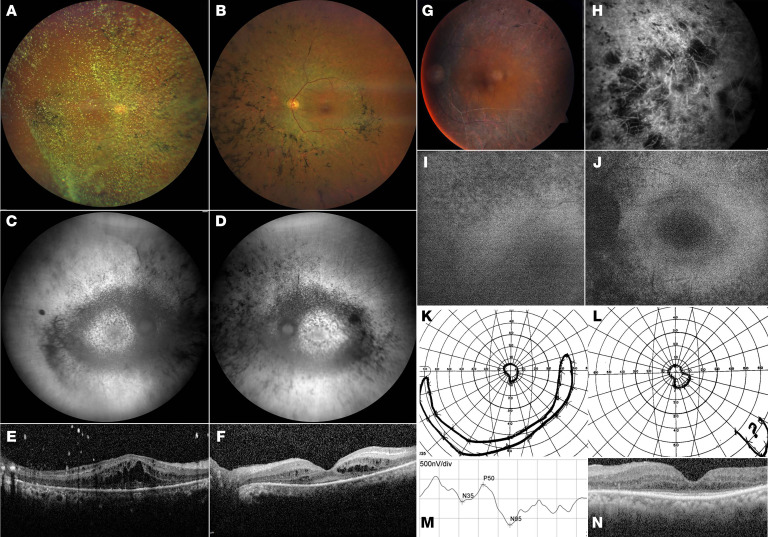
Defects in primary or motile cilia result in a variety of human pathologies, and retinal degeneration is frequently associated with these so-called ciliopathies. We found that homozygosity for a truncating variant in CEP162, a centrosome and microtubule-associated protein required for transition zone assembly during ciliogenesis and neuronal differentiation in the retina, caused late-onset retinitis pigmentosa in 2 unrelated families. The mutant CEP162-E646R*5 protein was expressed and properly localized to the mitotic spindle, but it was missing from the basal body in primary and photoreceptor cilia. This impaired recruitment of transition zone components to the basal body and corresponded to complete loss of CEP162 function at the ciliary compartment, reflected by delayed formation of dysmorphic cilia. In contrast, shRNA knockdown of Cep162 in the developing mouse retina increased cell death, which was rescued by expression of CEP162-E646R*5, indicating that the mutant retains its role for retinal neurogenesis. Human retinal degeneration thus resulted from specific loss of the ciliary function of CEP162.
Keywords: Cell Biology; Genetic diseases; Genetics; Molecular genetics; Retinopathy.
Figures





References
-
- Abramowicz I, et al. Oral-facial-digital syndrome type I cells exhibit impaired DNA repair; unanticipated consequences of defective OFD1 outside of the cilia network. Hum Mol Genet. 2017;26(1):19–32. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
