

A spinal muscular atrophy modifier implicates the SMN protein in SNARE complex assembly at neuromuscular synapses
- PMID: 36863345
- PMCID: PMC10164130
- DOI: 10.1016/j.neuron.2023.02.004
A spinal muscular atrophy modifier implicates the SMN protein in SNARE complex assembly at neuromuscular synapses
Abstract
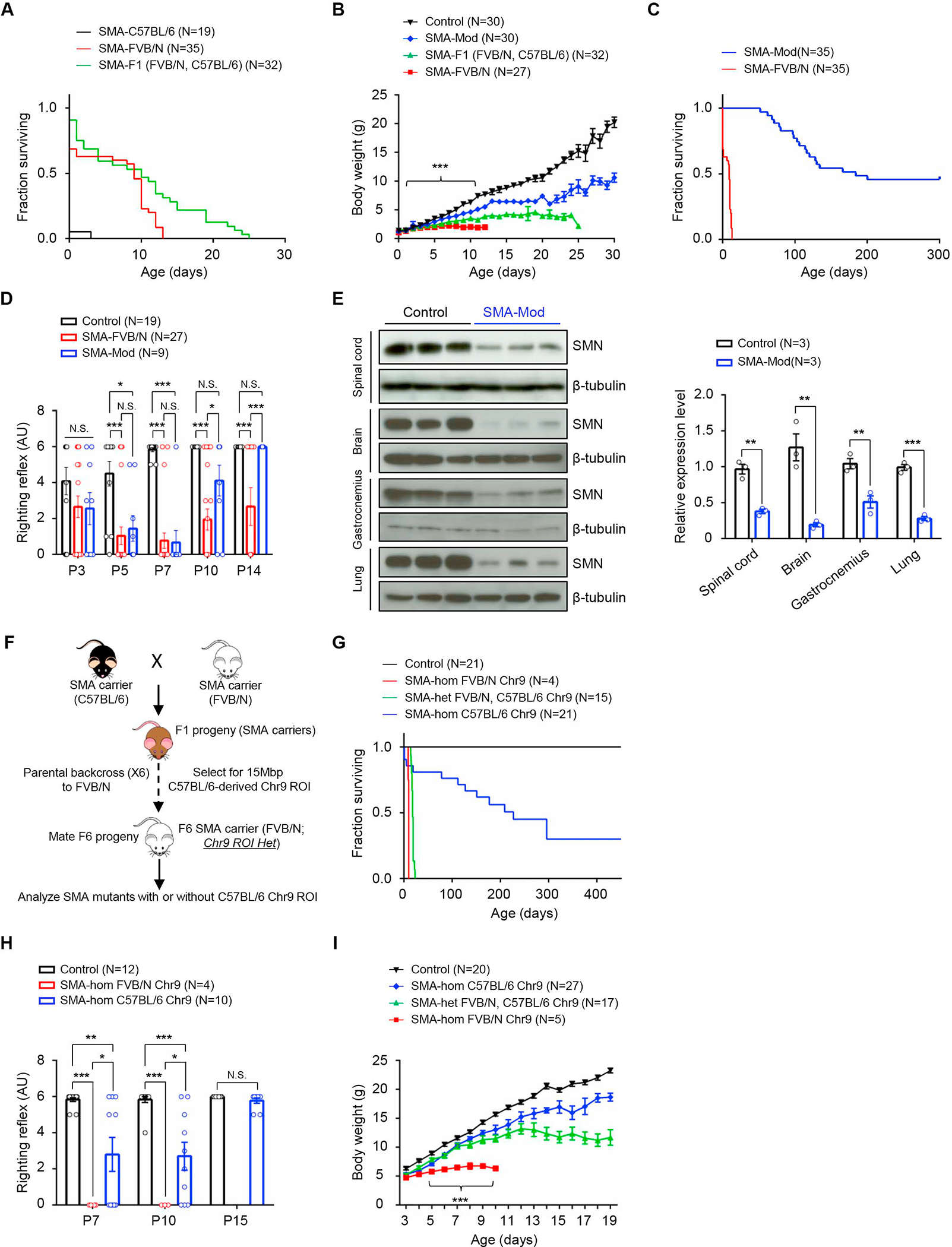
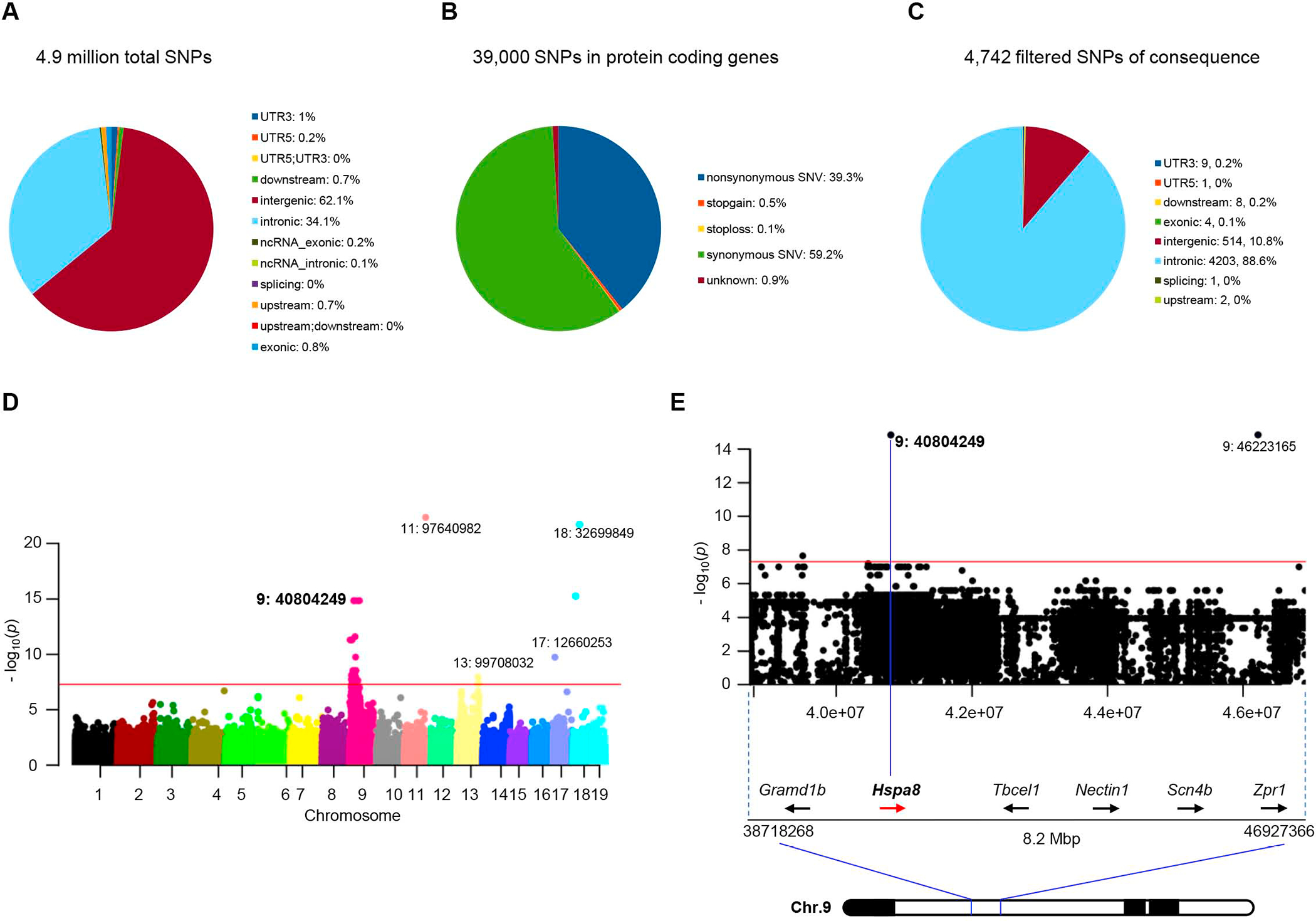
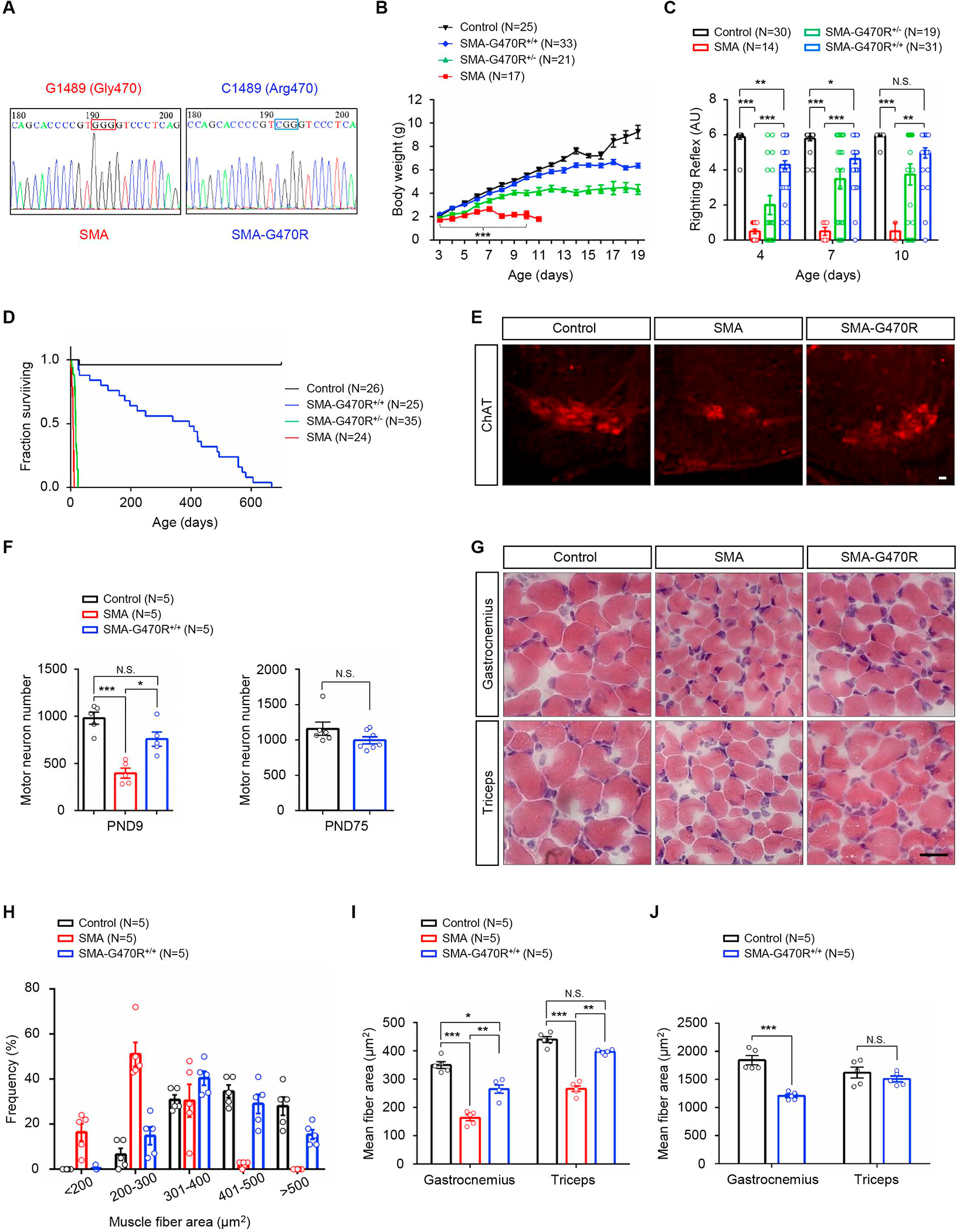
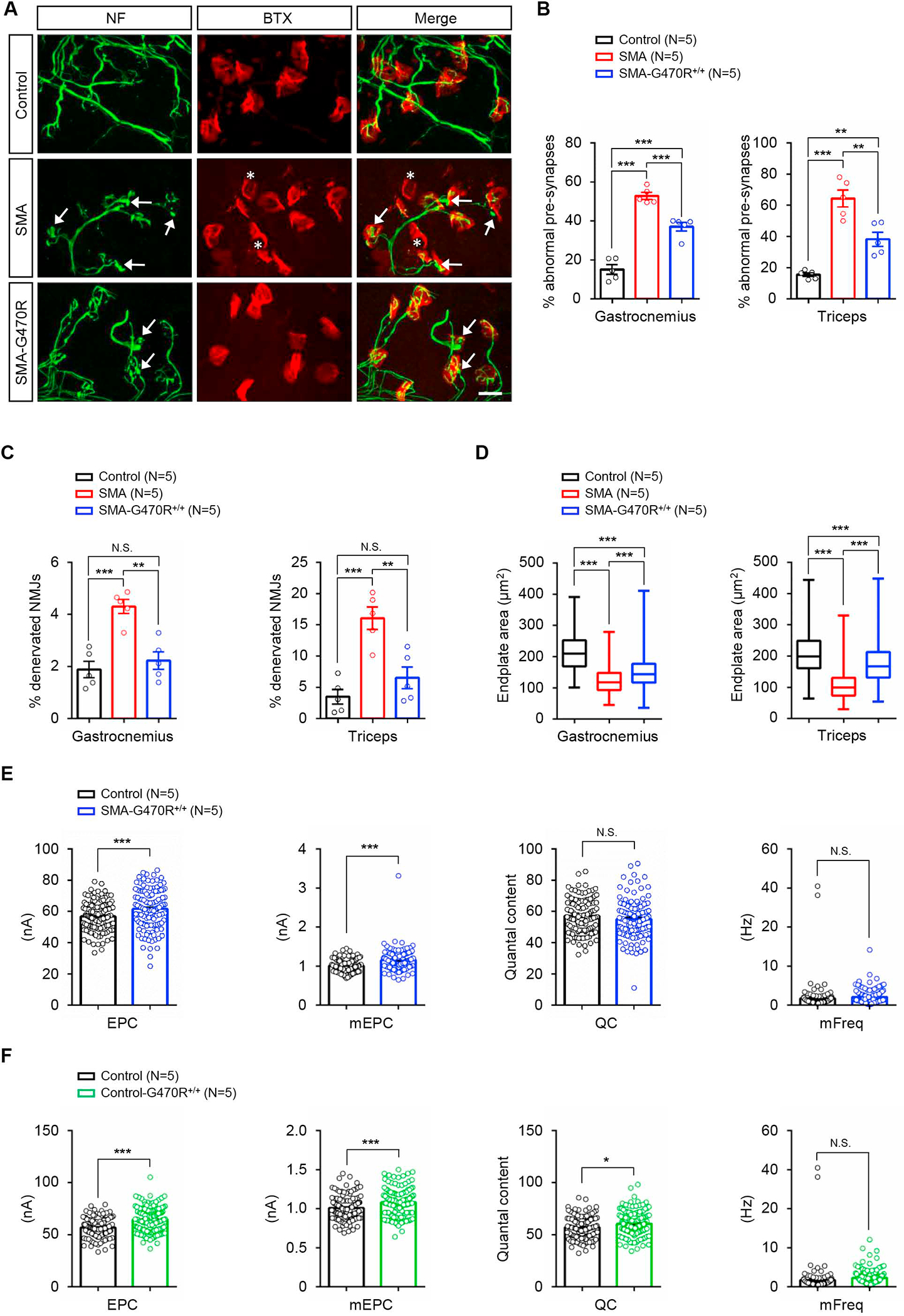
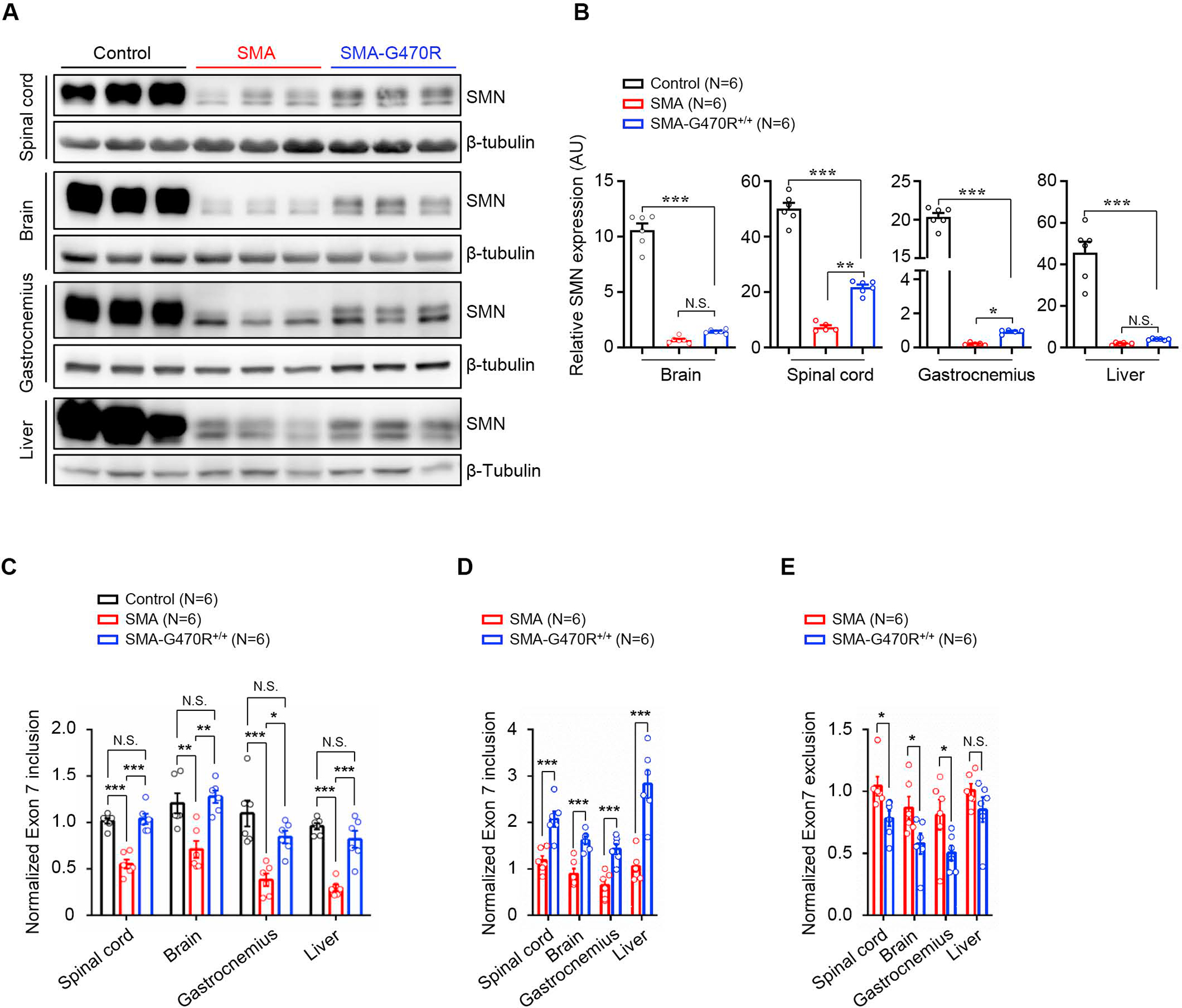
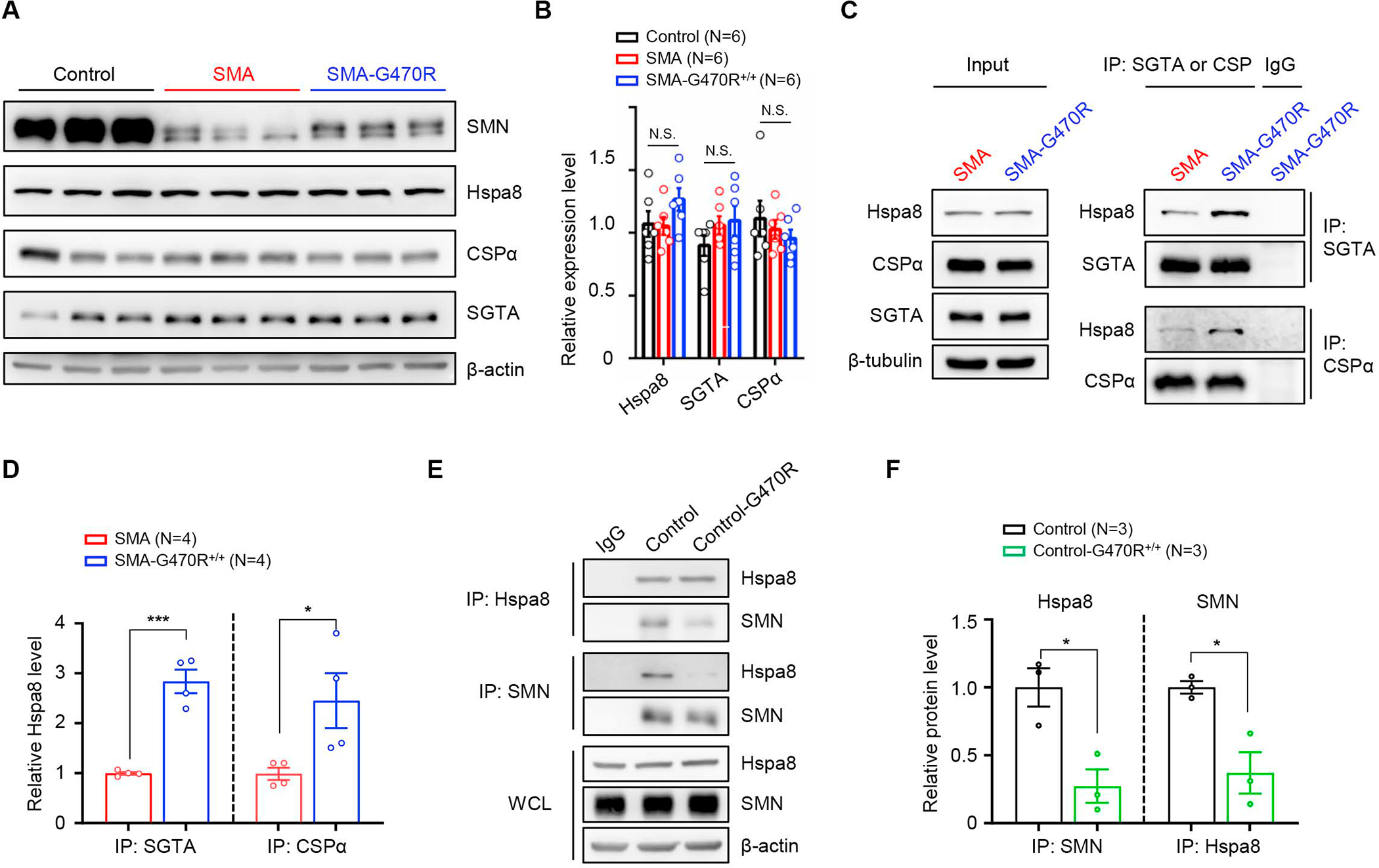
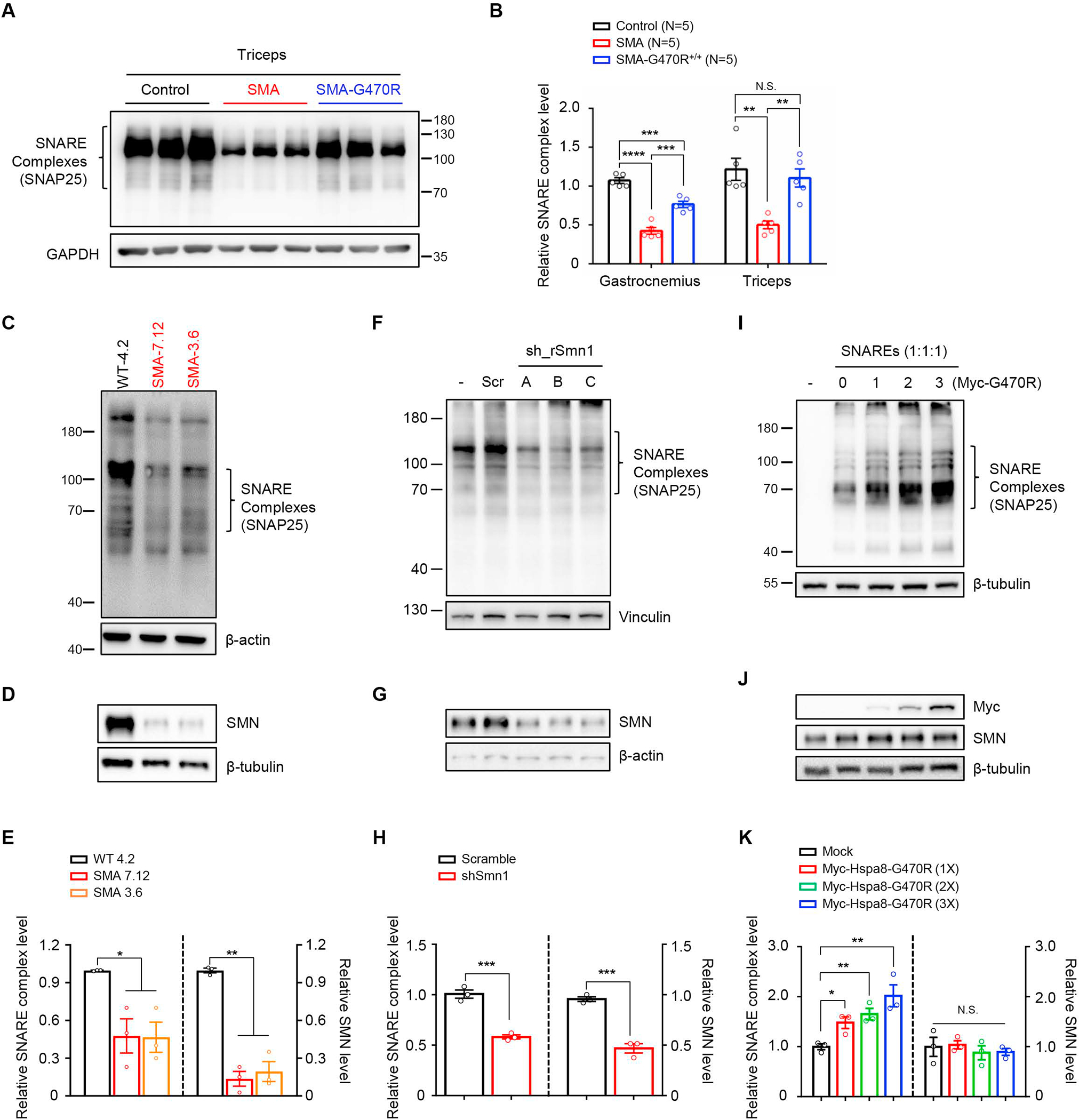
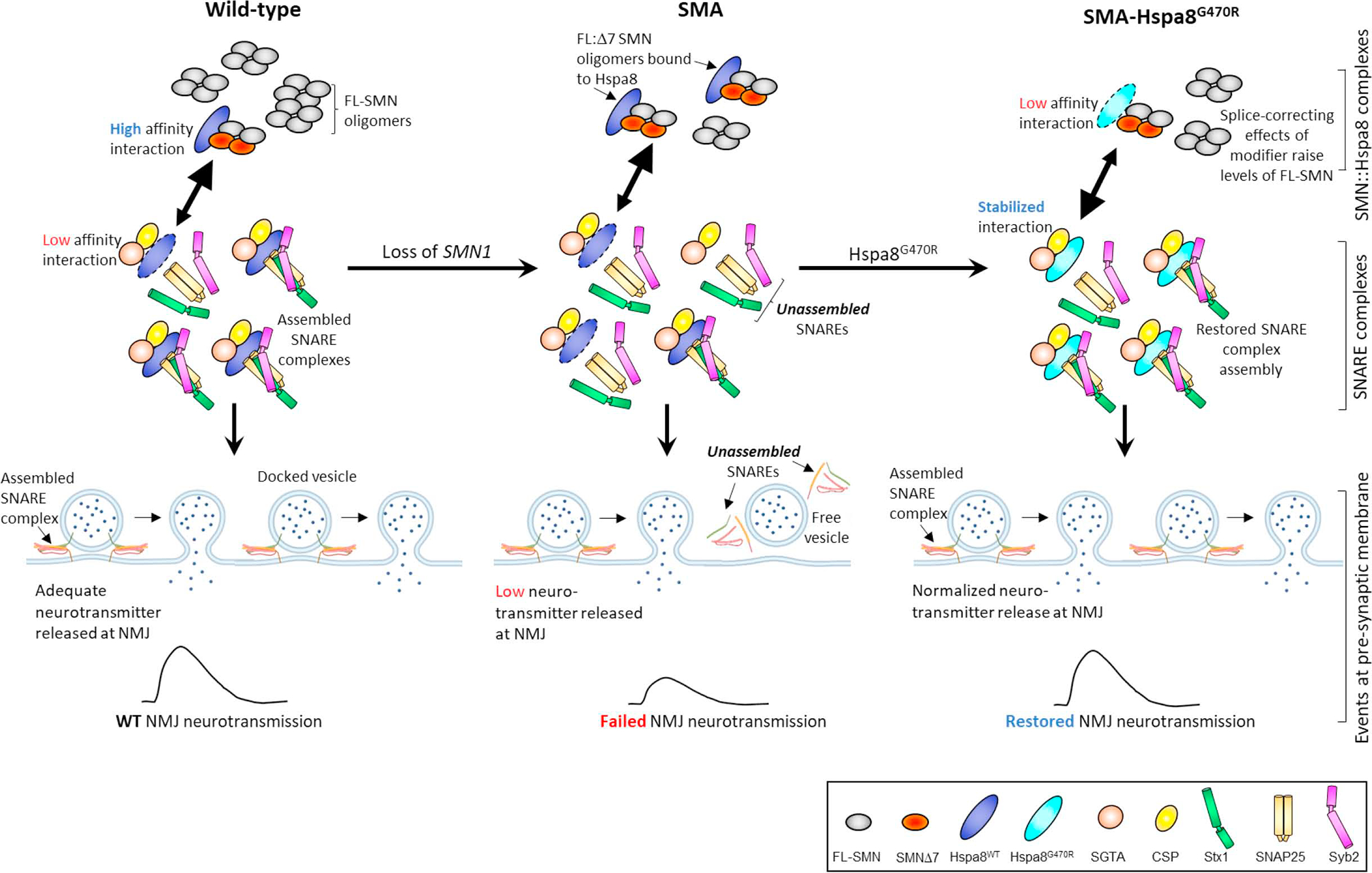
Reduced survival motor neuron (SMN) protein triggers the motor neuron disease, spinal muscular atrophy (SMA). Restoring SMN prevents disease, but it is not known how neuromuscular function is preserved. We used model mice to map and identify an Hspa8G470R synaptic chaperone variant, which suppressed SMA. Expression of the variant in the severely affected mutant mice increased lifespan >10-fold, improved motor performance, and mitigated neuromuscular pathology. Mechanistically, Hspa8G470R altered SMN2 splicing and simultaneously stimulated formation of a tripartite chaperone complex, critical for synaptic homeostasis, by augmenting its interaction with other complex members. Concomitantly, synaptic vesicular SNARE complex formation, which relies on chaperone activity for sustained neuromuscular synaptic transmission, was found perturbed in SMA mice and patient-derived motor neurons and was restored in modified mutants. Identification of the Hspa8G470R SMA modifier implicates SMN in SNARE complex assembly and casts new light on how deficiency of the ubiquitous protein causes motor neuron disease.
Keywords: Hspa8; SNARE complex assembly; modifiers; spinal muscular atrophy; survival motor neuron protein.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests J.-K.K. and U.R.M. are inventors on a provisional patent application filed by Columbia University on the use of Hspa8 as a means of treating neurodegenerative disease.
Figures
Comment in
-
SMN linked to SNARE complex assembly.Nat Rev Neurosci. 2023 May;24(5):267. doi: 10.1038/s41583-023-00694-w. Nat Rev Neurosci. 2023. PMID: 36932212 No abstract available.
-
An Hspa8 variant is a shocking modifier of spinal muscular atrophy in mice.Neuron. 2023 May 3;111(9):1349-1350. doi: 10.1016/j.neuron.2023.03.025. Neuron. 2023. PMID: 37141858
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
