

Analysis of structural variation among inbred mouse strains
- PMID: 36864393
- PMCID: PMC9983223
- DOI: 10.1186/s12864-023-09197-5
Analysis of structural variation among inbred mouse strains
Abstract
Background: 'Long read' sequencing methods have been used to identify previously uncharacterized structural variants that cause human genetic diseases. Therefore, we investigated whether long read sequencing could facilitate genetic analysis of murine models for human diseases.
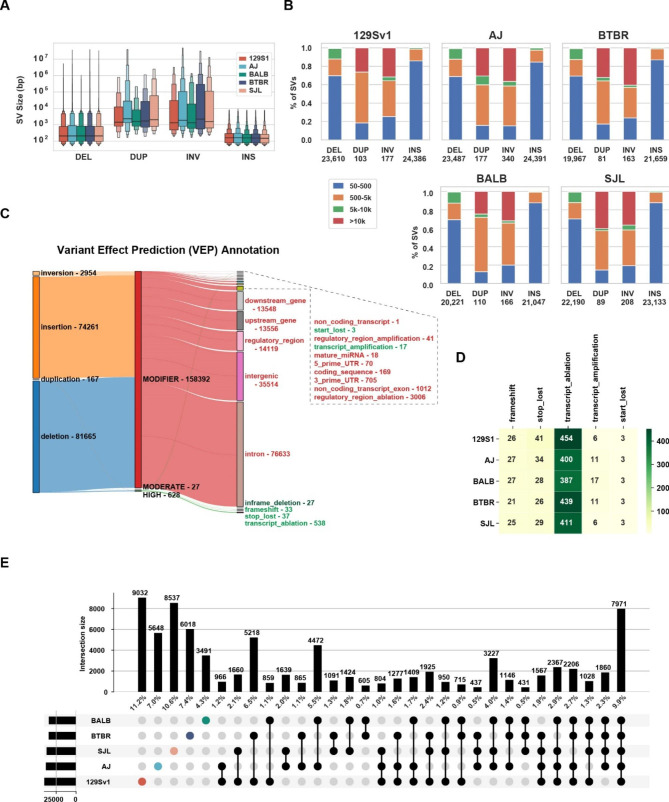
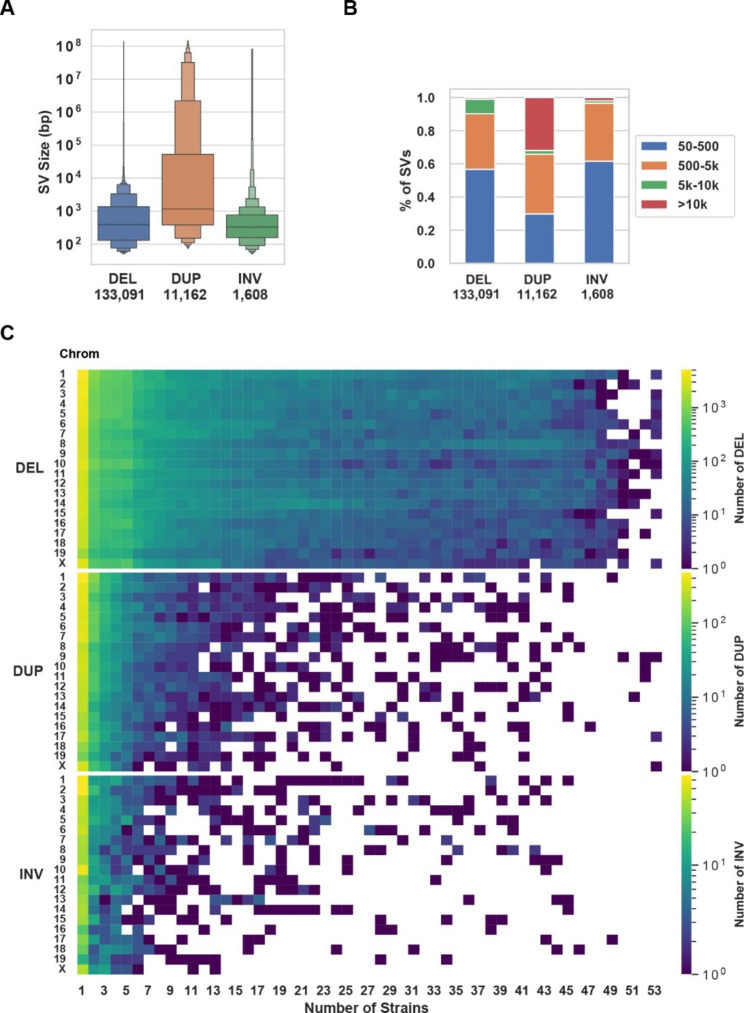
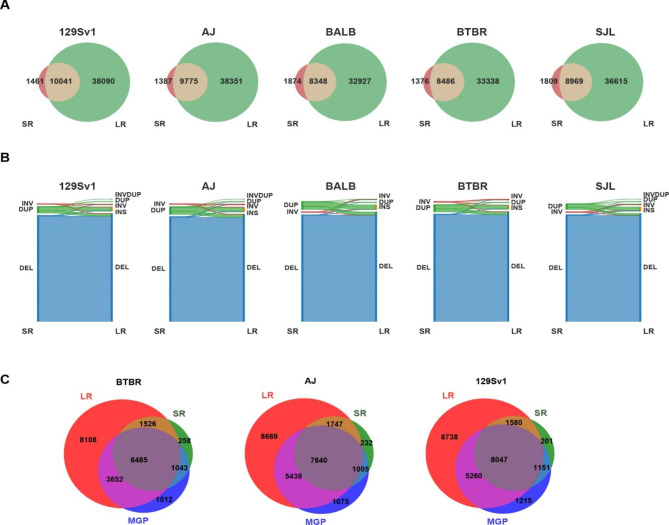
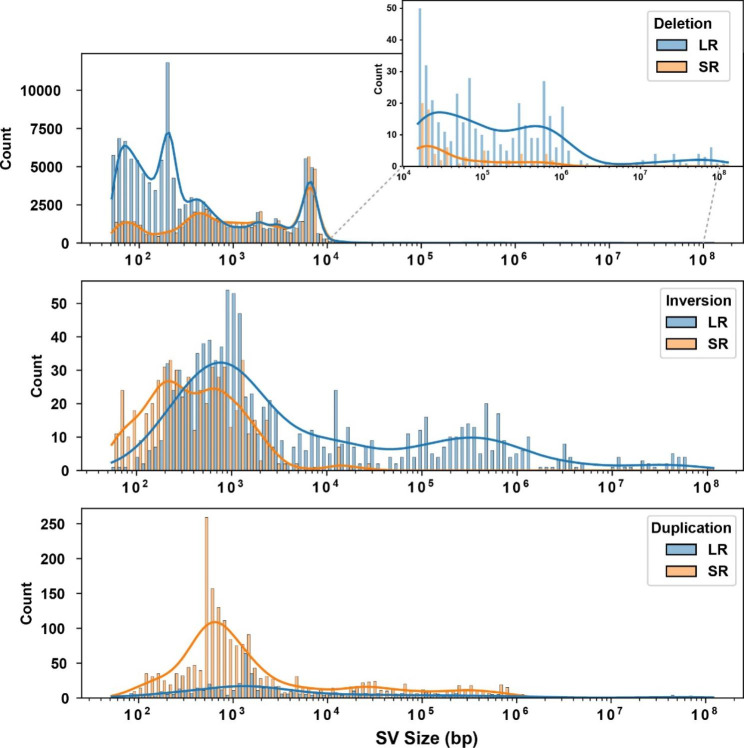
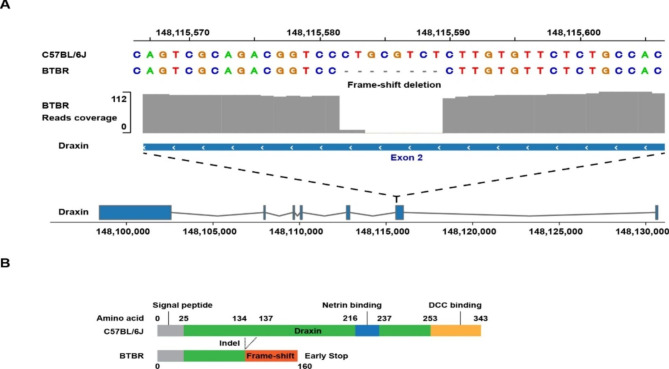
Results: The genomes of six inbred strains (BTBR T + Itpr3tf/J, 129Sv1/J, C57BL/6/J, Balb/c/J, A/J, SJL/J) were analyzed using long read sequencing. Our results revealed that (i) Structural variants are very abundant within the genome of inbred strains (4.8 per gene) and (ii) that we cannot accurately infer whether structural variants are present using conventional short read genomic sequence data, even when nearby SNP alleles are known. The advantage of having a more complete map was demonstrated by analyzing the genomic sequence of BTBR mice. Based upon this analysis, knockin mice were generated and used to characterize a BTBR-unique 8-bp deletion within Draxin that contributes to the BTBR neuroanatomic abnormalities, which resemble human autism spectrum disorder.
Conclusion: A more complete map of the pattern of genetic variation among inbred strains, which is produced by long read genomic sequencing of the genomes of additional inbred strains, could facilitate genetic discovery when murine models of human diseases are analyzed.
Keywords: Genetic analysis; Mouse genetic models; Structural variation.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
-
- Reiner J, Pisani L, Qiao W, Singh R, Yang Y, Shi L, Khan WA, Sebra R, Cohen N, Babu A, et al. Cytogenomic identification and long-read single molecule real-time (SMRT) sequencing of a Bardet-Biedl syndrome 9 (BBS9) deletion. NPJ Genom Med. 2018;3:3. doi: 10.1038/s41525-017-0042-3. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
