

This is a preprint.
FALCON systematically interrogates free fatty acid biology and identifies a novel mediator of lipotoxicity
- PMID: 36865221
- PMCID: PMC9979987
- DOI: 10.1101/2023.02.19.529127
FALCON systematically interrogates free fatty acid biology and identifies a novel mediator of lipotoxicity
Update in
-
FALCON systematically interrogates free fatty acid biology and identifies a novel mediator of lipotoxicity.Cell Metab. 2023 May 2;35(5):887-905.e11. doi: 10.1016/j.cmet.2023.03.018. Epub 2023 Apr 18. Cell Metab. 2023. PMID: 37075753 Free PMC article.
Abstract
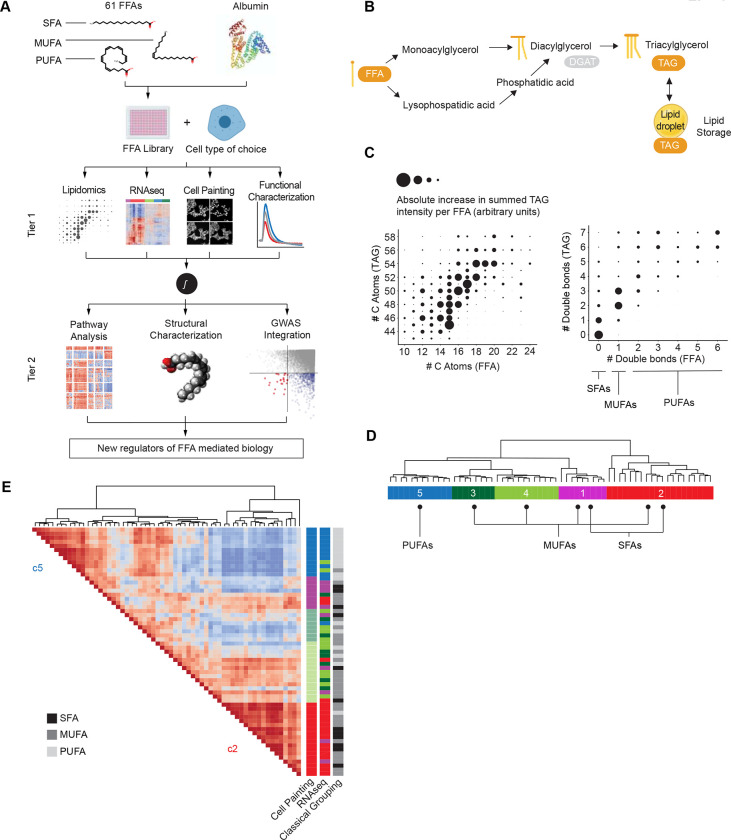
Cellular exposure to free fatty acids (FFA) is implicated in the pathogenesis of obesity-associated diseases. However, studies to date have assumed that a few select FFAs are representative of broad structural categories, and there are no scalable approaches to comprehensively assess the biological processes induced by exposure to diverse FFAs circulating in human plasma. Furthermore, assessing how these FFA- mediated processes interact with genetic risk for disease remains elusive. Here we report the design and implementation of FALCON (Fatty Acid Library for Comprehensive ONtologies) as an unbiased, scalable and multimodal interrogation of 61 structurally diverse FFAs. We identified a subset of lipotoxic monounsaturated fatty acids (MUFAs) with a distinct lipidomic profile associated with decreased membrane fluidity. Furthermore, we developed a new approach to prioritize genes that reflect the combined effects of exposure to harmful FFAs and genetic risk for type 2 diabetes (T2D). Importantly, we found that c-MAF inducing protein (CMIP) protects cells from exposure to FFAs by modulating Akt signaling and we validated the role of CMIP in human pancreatic beta cells. In sum, FALCON empowers the study of fundamental FFA biology and offers an integrative approach to identify much needed targets for diverse diseases associated with disordered FFA metabolism.
Highlights: FALCON (Fatty Acid Library for Comprehensive ONtologies) enables multimodal profiling of 61 free fatty acids (FFAs) to reveal 5 FFA clusters with distinct biological effectsFALCON is applicable to many and diverse cell typesA subset of monounsaturated FAs (MUFAs) equally or more toxic than canonical lipotoxic saturated FAs (SFAs) leads to decreased membrane fluidityNew approach prioritizes genes that represent the combined effects of environmental (FFA) exposure and genetic risk for diseaseC-Maf inducing protein (CMIP) is identified as a suppressor of FFA-induced lipotoxicity via Akt-mediated signaling.
Conflict of interest statement
Competing Interests
NW, JCF and AG are co-inventors of a patent on the composition, method and use for FFA screening, application No.: 52199-550P01US. A.G. serves as a founding advisor to a new company launched by Atlas Ventures, an agreement reviewed and managed by Brigham and Women’s Hospital, Mass General Brigham, and the Broad Institute of MIT and Harvard in accordance with their conflict of interest policies.
Figures







References
-
- Abifadel Marianne, Varret Mathilde, Rabès Jean-Pierre, Allard Delphine, Ouguerram Khadija, Devillers Martine, Cruaud Corinne, et al. 2003. “Mutations in PCSK9 Cause Autosomal Dominant Hypercholesterolemia.” Nature Genetics 34 (2): 154–56. - PubMed
-
- Al-Sulaiti Haya, Diboun Ilhame, Banu Sameem, Al-Emadi Mohamed, Amani Parvaneh, Harvey Thomas M., Dömling Alex S., Latiff Aishah, and Elrayess Mohamed A.. 2018. “Triglyceride Profiling in Adipose Tissues from Obese Insulin Sensitive, Insulin Resistant and Type 2 Diabetes Mellitus Individuals.” Journal of Translational Medicine 16 (1): 175. - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous