

This is a preprint.
Cerebellar Granule Cells Develop Non-neuronal 3D Genome Architecture over the Lifespan
- PMID: 36865235
- PMCID: PMC9980173
- DOI: 10.1101/2023.02.25.530020
Cerebellar Granule Cells Develop Non-neuronal 3D Genome Architecture over the Lifespan
Update in
-
Lifelong restructuring of 3D genome architecture in cerebellar granule cells.Science. 2023 Sep 8;381(6662):1112-1119. doi: 10.1126/science.adh3253. Epub 2023 Sep 7. Science. 2023. PMID: 37676945 Free PMC article.
Abstract
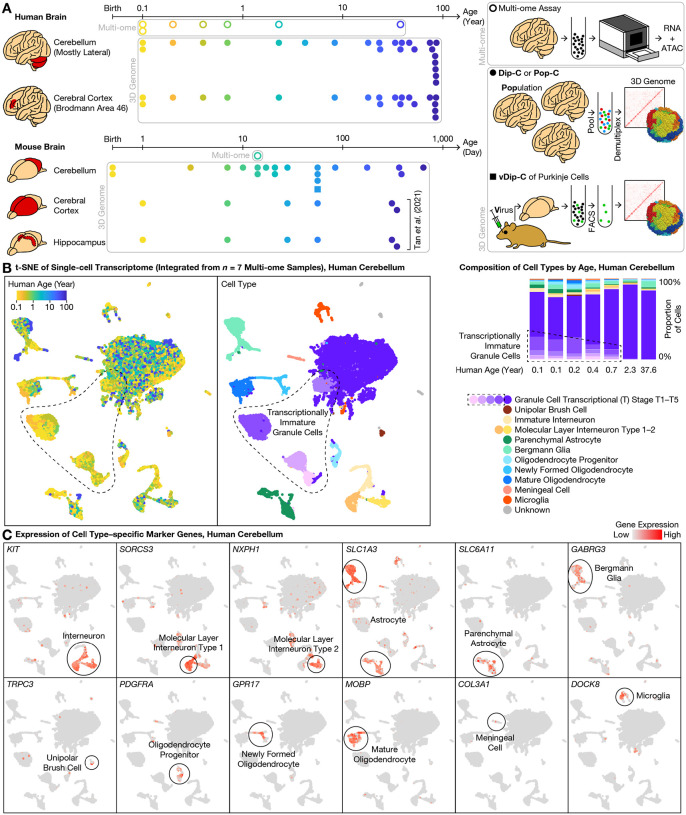
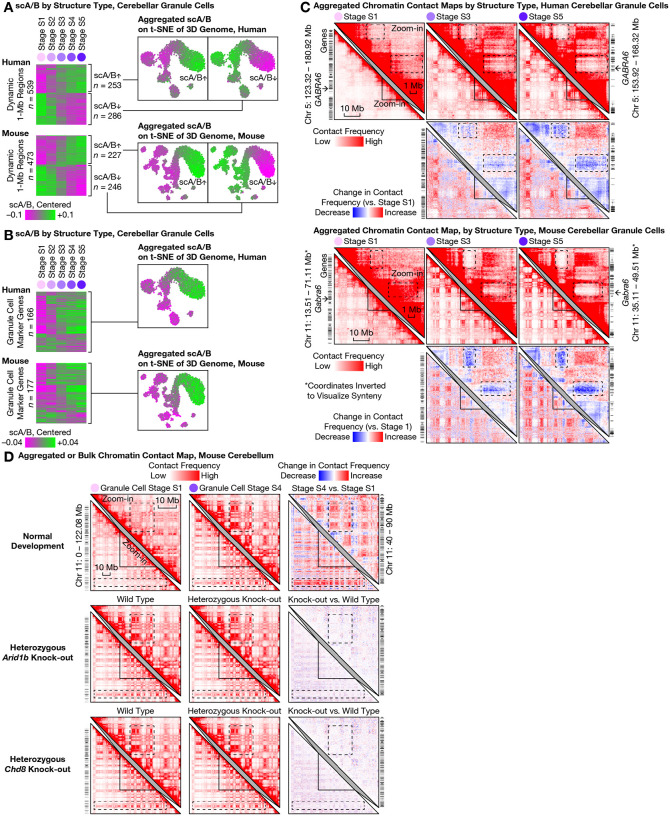
The cerebellum contains most of the neurons in the human brain, and exhibits unique modes of development, malformation, and aging. For example, granule cells-the most abundant neuron type-develop unusually late and exhibit unique nuclear morphology. Here, by developing our high-resolution single-cell 3D genome assay Dip-C into population-scale (Pop-C) and virus-enriched (vDip-C) modes, we were able to resolve the first 3D genome structures of single cerebellar cells, create life-spanning 3D genome atlases for both human and mouse, and jointly measure transcriptome and chromatin accessibility during development. We found that while the transcriptome and chromatin accessibility of human granule cells exhibit a characteristic maturation pattern within the first year of postnatal life, 3D genome architecture gradually remodels throughout life into a non-neuronal state with ultra-long-range intra-chromosomal contacts and specific inter-chromosomal contacts. This 3D genome remodeling is conserved in mice, and robust to heterozygous deletion of chromatin remodeling disease-associated genes (Chd8 or Arid1b). Together these results reveal unexpected and evolutionarily-conserved molecular processes underlying the unique development and aging of the mammalian cerebellum.
Conflict of interest statement
Competing interests: LT is an inventor on a patent application US16/615,872 filed by Harvard University that covers Dip-C.
Figures



References
-
- Chi Y., Shi J., Xing D., Tan L., Every gene everywhere all at once: High-precision measurement of 3D chromosome architecture with single-cell Hi-C. Frontiers in Molecular Biosciences. 9 (2022) (available at https://www.frontiersin.org/articles/10.3389/fmolb.2022.959688). - DOI - PMC - PubMed
-
- Tan L., Xing D., Daley N., Xie X. S., Three-dimensional genome structures of single sensory neurons in mouse visual and olfactory systems. Nat Struct Mol Biol. 26, 297–307 (2019). - PubMed
-
- Tan L., Ma W., Wu H., Zheng Y., Xing D., Chen R., Li X., Daley N., Deisseroth K., Xie X. S., Changes in genome architecture and transcriptional dynamics progress independently of sensory experience during post-natal brain development. Cell. 184, 741–758.e17 (2021). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources