

cAMP-PDE signaling in COPD: Review of cellular, molecular and clinical features
- PMID: 36865738
- PMCID: PMC9971187
- DOI: 10.1016/j.bbrep.2023.101438
cAMP-PDE signaling in COPD: Review of cellular, molecular and clinical features
Abstract
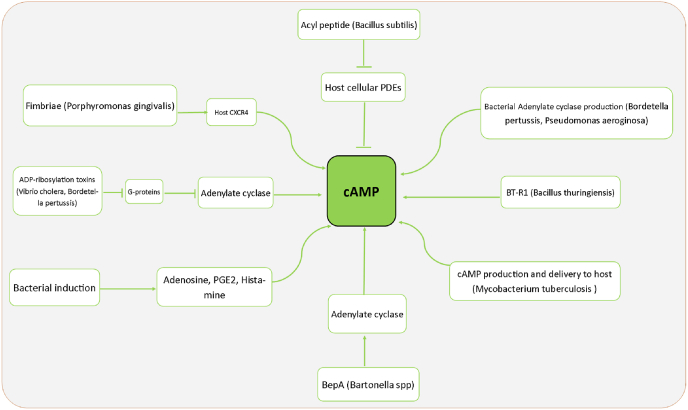
Chronic obstructive pulmonary disease (COPD) is the fourth leading cause of death among non-contagious diseases in the world. PDE inhibitors are among current medicines prescribed for COPD treatment of which, PDE-4 family is the predominant PDE isoform involved in hydrolyzing cyclic adenosine monophosphate (cAMP) that regulates the inflammatory responses in neutrophils, lymphocytes, macrophages and epithelial cells The aim of this study is to investigate the cellular and molecular mechanisms of cAMP-PDE signaling, as an important pathway in the treatment management of patients with COPD. In this review, a comprehensive literature review was performed about the effect of PDEs in COPD. Generally, PDEs are overexpressed in COPD patients, resulting in cAMP inactivation and decreased cAMP hydrolysis from AMP. At normal amounts, cAMP is one of the essential agents in regulating metabolism and suppressing inflammatory responses. Low amount of cAMP lead to activation of downstream inflammatory signaling pathways. PDE4 and PDE7 mRNA transcript levels were not altered in polymorphonuclear leukocytes and CD8 lymphocytes originating from the peripheral venous blood of stable COPD subjects compared to healthy controls. Therefore, cAMP-PDE signaling pathway is one of the most important signaling pathways involved in COPD. By examining the effects of different drugs in this signaling pathway critical steps can be taken in the treatment of this disease.
Keywords: COPD Therapy; Phosphodiesterase inhibitors; cAMP signaling.
© 2023 The Authors.
Conflict of interest statement
Authors have no conflict of interest to declare.
Figures




References
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
