

Robust total X-ray scattering workflow to study correlated motion of proteins in crystals
- PMID: 36869043
- PMCID: PMC9984388
- DOI: 10.1038/s41467-023-36734-3
Robust total X-ray scattering workflow to study correlated motion of proteins in crystals
Abstract
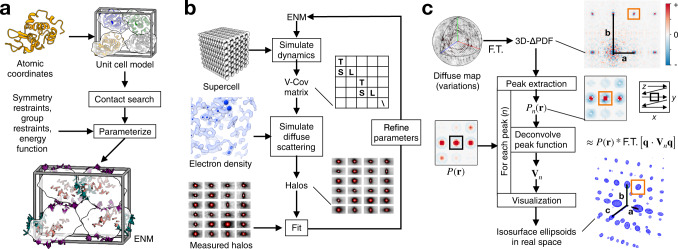
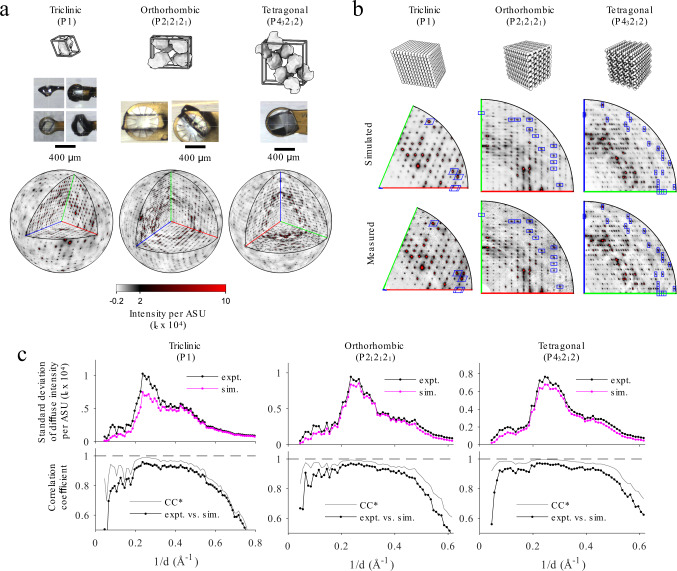
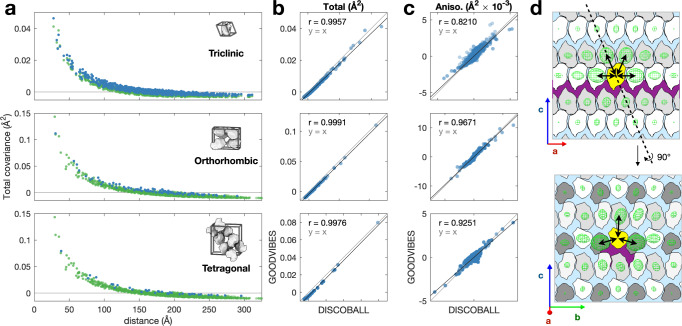
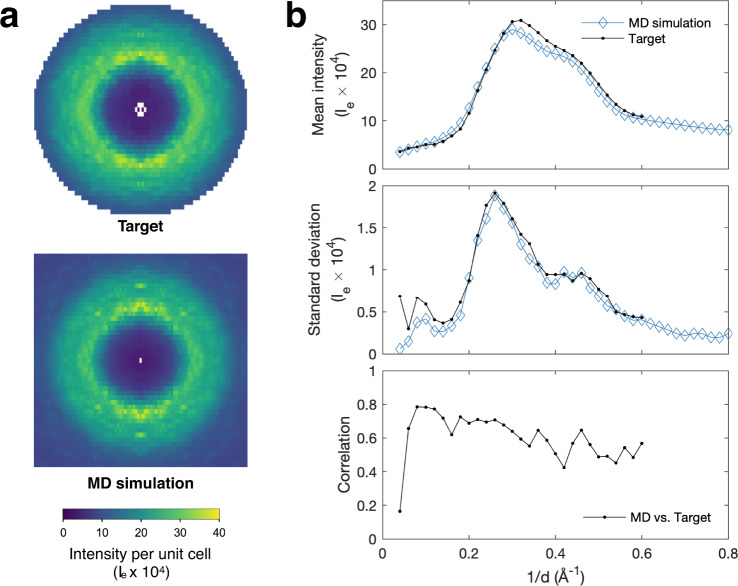
The breathing motions of proteins are thought to play a critical role in function. However, current techniques to study key collective motions are limited to spectroscopy and computation. We present a high-resolution experimental approach based on the total scattering from protein crystals at room temperature (TS/RT-MX) that captures both structure and collective motions. To reveal the scattering signal from protein motions, we present a general workflow that enables robust subtraction of lattice disorder. The workflow introduces two methods: GOODVIBES, a detailed and refinable lattice disorder model based on the rigid-body vibrations of a crystalline elastic network; and DISCOBALL, an independent method of validation that estimates the displacement covariance between proteins in the lattice in real space. Here, we demonstrate the robustness of this workflow and further demonstrate how it can be interfaced with MD simulations towards obtaining high-resolution insight into functionally important protein motions.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


Similar articles
-
Inelastic X-ray scattering studies of the short-time collective vibrational motions in hydrated lysozyme powders and their possible relation to enzymatic function.J Phys Chem B. 2013 Jan 31;117(4):1186-95. doi: 10.1021/jp312842m. Epub 2013 Jan 18. J Phys Chem B. 2013. PMID: 23301848
-
Liquid-like and rigid-body motions in molecular-dynamics simulations of a crystalline protein.Struct Dyn. 2019 Dec 18;6(6):064704. doi: 10.1063/1.5132692. eCollection 2019 Nov. Struct Dyn. 2019. PMID: 31867408 Free PMC article.
-
NMR study of cation dynamics in three crystalline states of 1-butyl-3-methylimidazolium hexafluorophosphate exhibiting crystal polymorphism.J Phys Chem B. 2012 Mar 29;116(12):3780-8. doi: 10.1021/jp300636s. Epub 2012 Mar 19. J Phys Chem B. 2012. PMID: 22380424
-
Correlated Motions in Structural Biology.Biochemistry. 2021 Aug 3;60(30):2331-2340. doi: 10.1021/acs.biochem.1c00420. Epub 2021 Jul 22. Biochemistry. 2021. PMID: 34291898 Free PMC article. Review.
-
Efficient characterization of collective motions and interresidue correlations in proteins by low-resolution simulations.Biochemistry. 1997 Nov 4;36(44):13512-23. doi: 10.1021/bi971611f. Biochemistry. 1997. PMID: 9354619 Review.
Cited by
-
3D variability analysis reveals a hidden conformational change controlling ammonia transport in human asparagine synthetase.Nat Commun. 2024 Dec 3;15(1):10538. doi: 10.1038/s41467-024-54912-9. Nat Commun. 2024. PMID: 39627226 Free PMC article.
-
Functional Protein Dynamics in a Crystal.bioRxiv [Preprint]. 2024 Mar 24:2023.07.06.548023. doi: 10.1101/2023.07.06.548023. bioRxiv. 2024. Update in: Nat Commun. 2024 Apr 15;15(1):3244. doi: 10.1038/s41467-024-47473-4. PMID: 37461732 Free PMC article. Updated. Preprint.
-
Scaling and merging macromolecular diffuse scattering with mdx2.Acta Crystallogr D Struct Biol. 2024 May 1;80(Pt 5):299-313. doi: 10.1107/S2059798324002705. Epub 2024 Apr 12. Acta Crystallogr D Struct Biol. 2024. PMID: 38606664 Free PMC article.
-
Reconciling ASPP-p53 binding mode discrepancies through an ensemble binding framework that bridges crystallography and NMR data.PLoS Comput Biol. 2024 Feb 7;20(2):e1011519. doi: 10.1371/journal.pcbi.1011519. eCollection 2024 Feb. PLoS Comput Biol. 2024. PMID: 38324587 Free PMC article.
-
Functional protein dynamics in a crystal.Nat Commun. 2024 Apr 15;15(1):3244. doi: 10.1038/s41467-024-47473-4. Nat Commun. 2024. PMID: 38622111 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
