

A noncanonical response to replication stress protects genome stability through ROS production, in an adaptive manner
- PMID: 36869180
- PMCID: PMC10154342
- DOI: 10.1038/s41418-023-01141-0
A noncanonical response to replication stress protects genome stability through ROS production, in an adaptive manner
Abstract
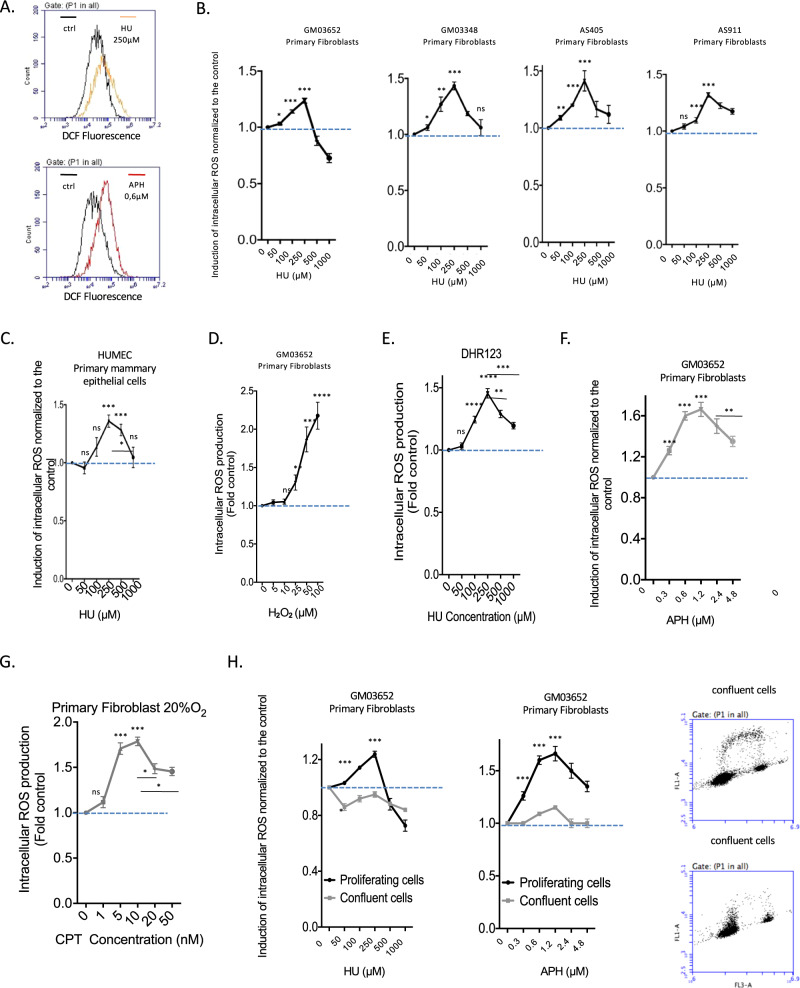
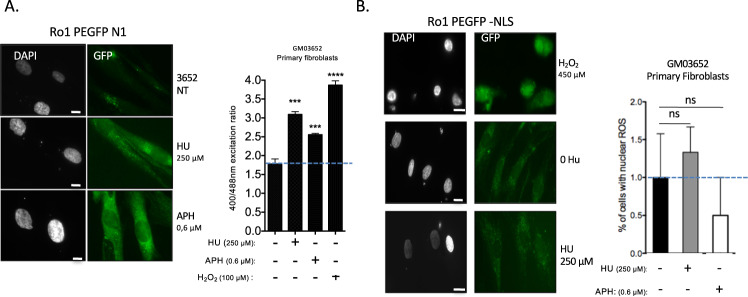
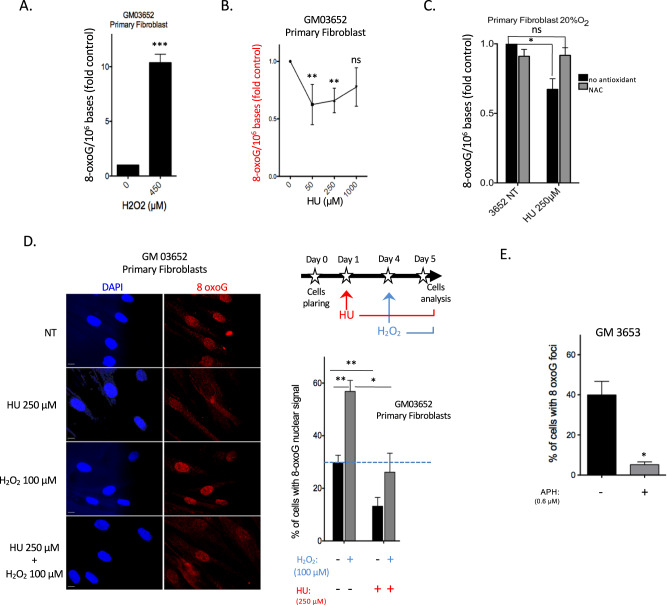
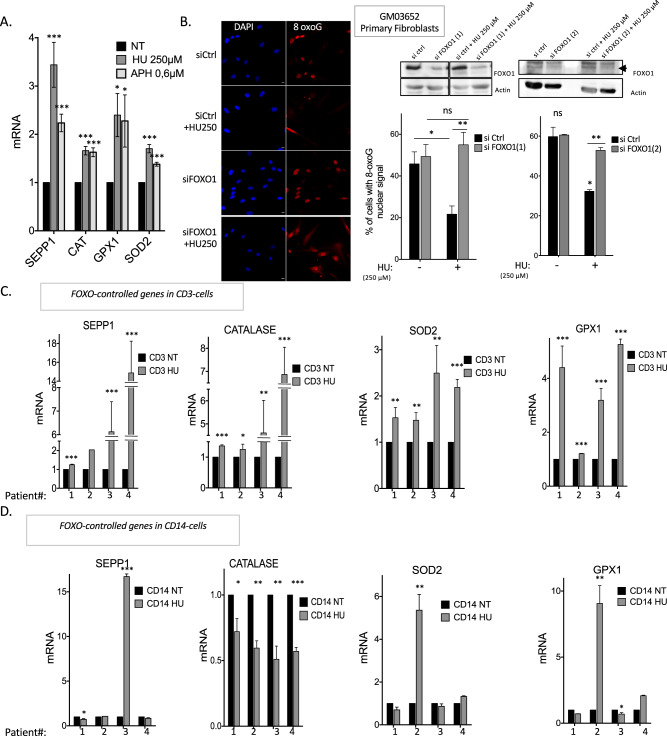
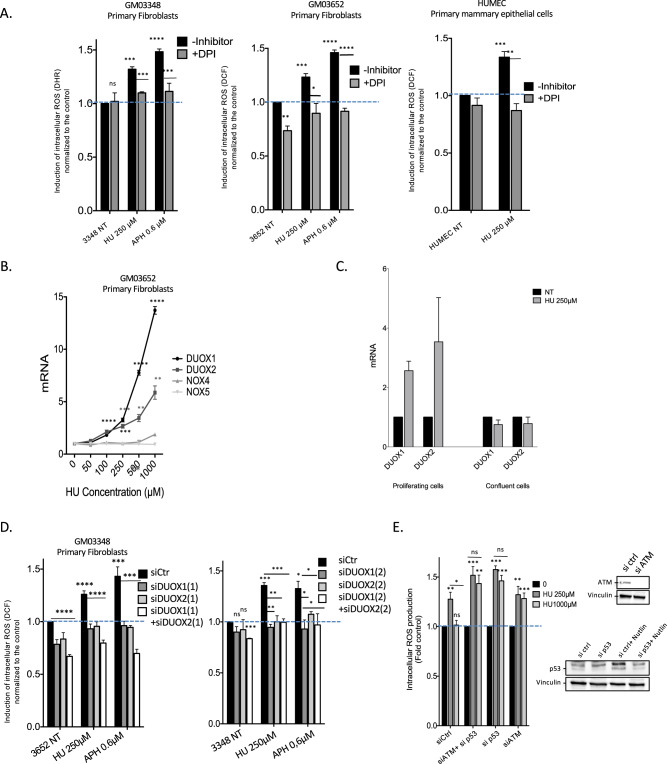
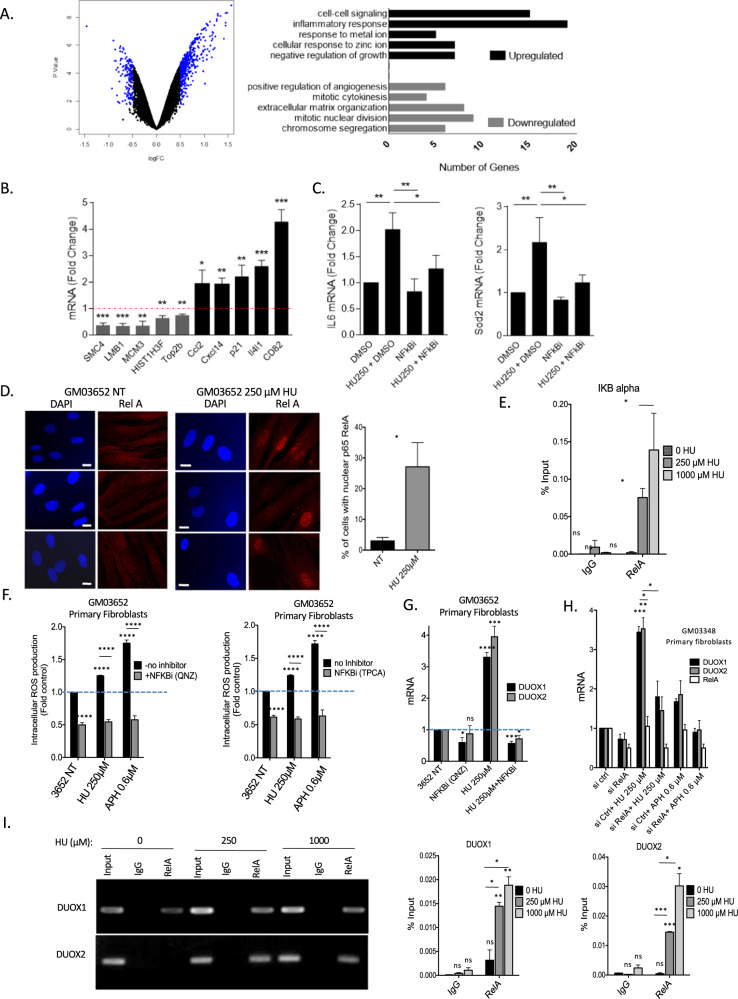
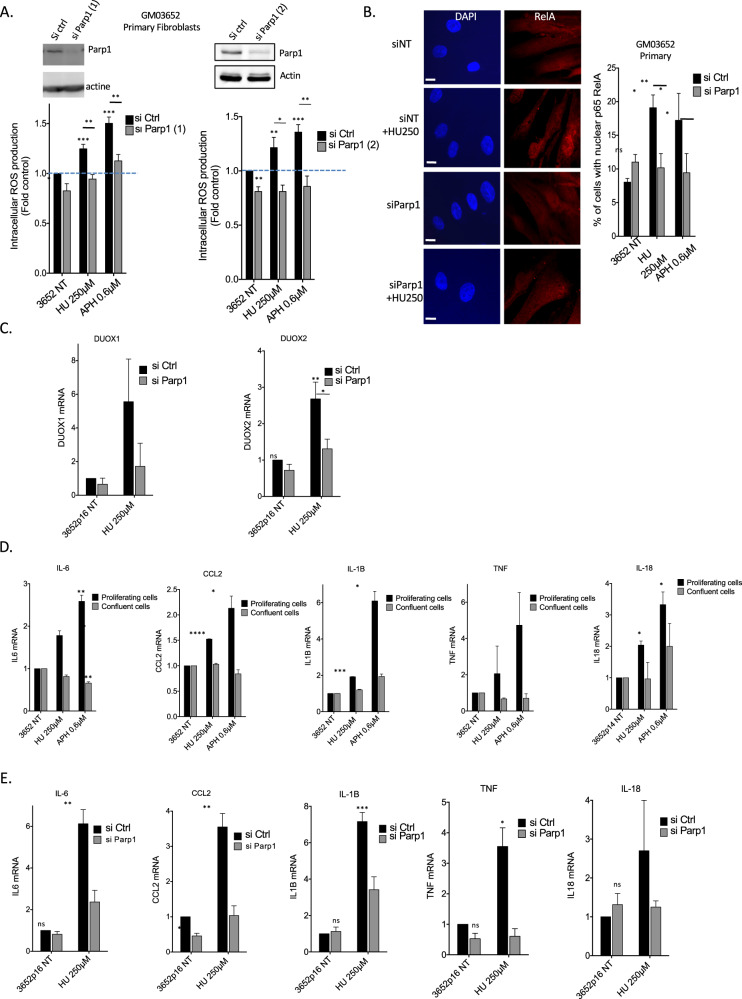
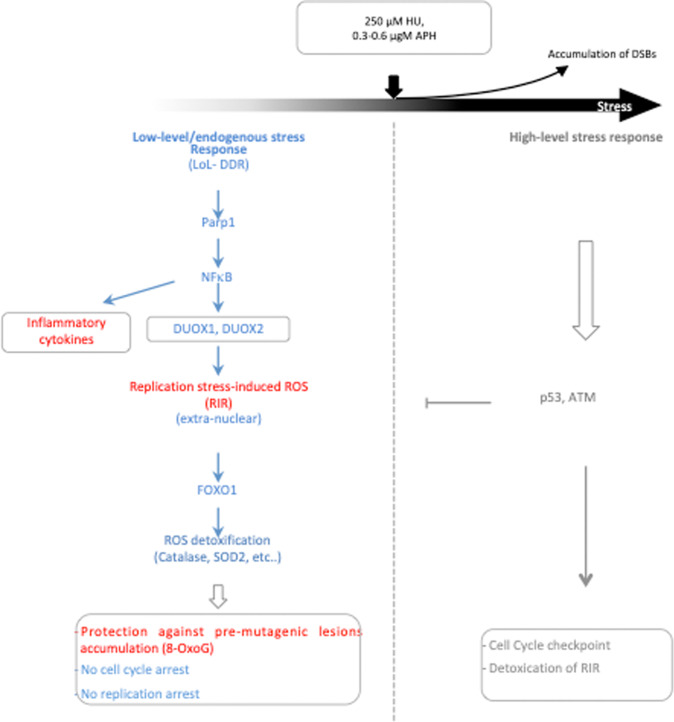
Cells are inevitably challenged by low-level/endogenous stresses that do not arrest DNA replication. Here, in human primary cells, we discovered and characterized a noncanonical cellular response that is specific to nonblocking replication stress. Although this response generates reactive oxygen species (ROS), it induces a program that prevents the accumulation of premutagenic 8-oxoguanine in an adaptive way. Indeed, replication stress-induced ROS (RIR) activate FOXO1-controlled detoxification genes such as SEPP1, catalase, GPX1, and SOD2. Primary cells tightly control the production of RIR: They are excluded from the nucleus and are produced by the cellular NADPH oxidases DUOX1/DUOX2, whose expression is controlled by NF-κB, which is activated by PARP1 upon replication stress. In parallel, inflammatory cytokine gene expression is induced through the NF-κB-PARP1 axis upon nonblocking replication stress. Increasing replication stress intensity accumulates DNA double-strand breaks and triggers the suppression of RIR by p53 and ATM. These data underline the fine-tuning of the cellular response to stress that protects genome stability maintenance, showing that primary cells adapt their responses to replication stress severity.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Specific Low/Endogenous Replication Stress Response Protects Genomic Stability via Controlled ROS Production in an Adaptive Way and Is Dysregulated in Transformed Cells.Cells. 2025 Jul 31;14(15):1183. doi: 10.3390/cells14151183. Cells. 2025. PMID: 40801615 Free PMC article. Review.
-
Dysregulation of the low-level replication stress response in transformed cell lines.Sci Rep. 2025 Jun 6;15(1):20013. doi: 10.1038/s41598-025-05172-0. Sci Rep. 2025. PMID: 40481239 Free PMC article.
-
Luteolin protects HUVECs from TNF-α-induced oxidative stress and inflammation via its effects on the Nox4/ROS-NF-κB and MAPK pathways.J Atheroscler Thromb. 2014;21(8):768-83. doi: 10.5551/jat.23697. Epub 2014 Mar 12. J Atheroscler Thromb. 2014. PMID: 24621786
-
High glucose induces renal mesangial cell proliferation and fibronectin expression through JNK/NF-κB/NADPH oxidase/ROS pathway, which is inhibited by resveratrol.Int J Biochem Cell Biol. 2012 Apr;44(4):629-38. doi: 10.1016/j.biocel.2012.01.001. Epub 2012 Jan 9. Int J Biochem Cell Biol. 2012. PMID: 22245600
-
MicroRNA Targeting Nicotinamide Adenine Dinucleotide Phosphate Oxidases in Cancer.Antioxid Redox Signal. 2020 Feb 10;32(5):267-284. doi: 10.1089/ars.2019.7918. Epub 2019 Nov 21. Antioxid Redox Signal. 2020. PMID: 31656079 Review.
Cited by
-
Simulating Space Conditions Evokes Different DNA Damage Responses in Immature and Mature Cells of the Human Hematopoietic System.Int J Mol Sci. 2023 Sep 6;24(18):13761. doi: 10.3390/ijms241813761. Int J Mol Sci. 2023. PMID: 37762064 Free PMC article.
-
Cellular Responses to Widespread DNA Replication Stress.Int J Mol Sci. 2023 Nov 29;24(23):16903. doi: 10.3390/ijms242316903. Int J Mol Sci. 2023. PMID: 38069223 Free PMC article. Review.
-
In vivo reduction of RAD51-mediated homologous recombination triggers aging but impairs oncogenesis.EMBO J. 2023 Oct 16;42(20):e110844. doi: 10.15252/embj.2022110844. Epub 2023 Sep 4. EMBO J. 2023. PMID: 37661798 Free PMC article.
-
Pamiparib as consolidation treatment after concurrent chemoradiotherapy of limited-stage small cell lung cancer: a single-arm, open-label phase 2 trial.Radiat Oncol. 2024 Apr 12;19(1):47. doi: 10.1186/s13014-024-02437-2. Radiat Oncol. 2024. PMID: 38610031 Free PMC article. Clinical Trial.
-
FoxO1-Overexpressed Small Extracellular Vesicles Derived from hPDLSCs Promote Periodontal Tissue Regeneration by Reducing Mitochondrial Dysfunction to Regulate Osteogenesis and Inflammation.Int J Nanomedicine. 2024 Aug 27;19:8751-8768. doi: 10.2147/IJN.S470419. eCollection 2024. Int J Nanomedicine. 2024. PMID: 39220194 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
