

Neuromuscular junction involvement in inherited motor neuropathies: genetic heterogeneity and effect of oral salbutamol treatment
- PMID: 36869887
- PMCID: PMC10188419
- DOI: 10.1007/s00415-023-11643-z
Neuromuscular junction involvement in inherited motor neuropathies: genetic heterogeneity and effect of oral salbutamol treatment
Abstract
Objectives: Inherited defects of the neuromuscular junction (NMJ) comprise an increasingly diverse range of diseases. Several recently identified genes highlight the overlap between peripheral neuropathies and congenital myasthenic syndromes (CMS). The beta-2 adrenergic receptor agonist salbutamol has been shown to provide symptomatic benefit in CMS, while improving structural defects at the NMJ. Based on these findings, we identified cases of motor neuropathy with NMJ dysfunction and assessed the effect of salbutamol on motor function.
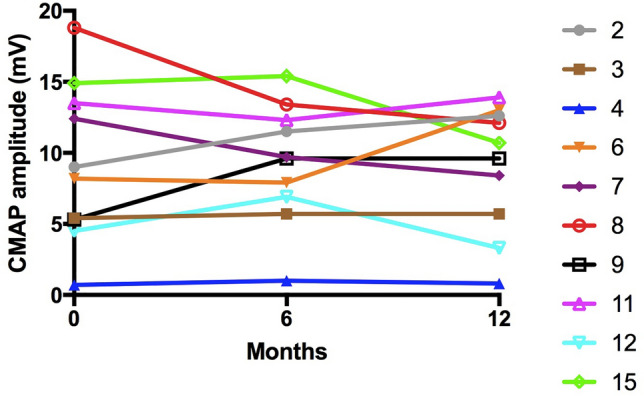
Methods: Cases of motor neuropathy with significant NMJ dysfunction, were identified using repetitive nerve stimulation and single fibre electromyography. Oral salbutamol was administered for 12 months. Repeat neurophysiological and clinical assessments were undertaken at baseline, 6 months and 12 months.
Results: Significant defects of neuromuscular transmission were identified in 15 patients harbouring a range of genetic defects, including mutations in GARS1, DNM2, SYT2 and DYNC1H. No clear benefit on motor function was seen following the administration of 12 months of oral salbutamol; however, there was a significant improvement in patient reported fatigue. In addition, no clear effect on neurophysiological parameters was seen in patients treated with salbutamol. Side-effects due to off-target beta-adrenergic effects were significant in the patient cohort.
Conclusion: These results highlight the involvement of the NMJ in several subtypes of motor neuropathies, including subtypes of neuropathy due to deficits in mitochondrial fusion-fission, synaptic vesicle transport, calcium channels and tRNA synthetases. Whether the NMJ dysfunction is simply due to muscle reinnervation or a pathology unrelated to denervation is unknown. The involvement of the NMJ may represent a novel therapeutic target in these conditions. However, treatment regimens will need to be more targeted for patients with primary inherited defects of neuromuscular transmission.
Keywords: Charcot-Marie-Tooth disease (CMT); Distal hereditary motor neuropathy (dHMN); Genetic defects; Inherited peripheral neuropathy; Neuromuscular junction (NMJ); Neuromuscular transmission (NMT).
© 2023. The Author(s).
Conflict of interest statement
The authors have no conflicts of interest and the publication has not been submitted to any other journal. Dr. Grace McMacken reports no disclosures. Dr. Roger Whittaker reports no disclosures. Ruth Wake reports no disclosures. Dr. Hanns Lochmüller reports no disclosures. Dr. Rita Horvath reports no disclosures. The Authors have no financial or non-financial interests that are directly or indirectly related to the work submitted for publication.
Figures


References
-
- Vavlitou N, Sargiannidou I, Markoullis K, Kyriacou K, Scherer SS, Kleopa KA. Axonal pathology precedes demyelination in a mouse model of X-linked demyelinating/ type I Charcot-Marie tooth (CMT1X) neuropathy. J Neuropathol Exp Neurol. 2010;69(9):945–958. doi: 10.1097/NEN.0b013e3181efa658. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
