

Development of the novel ACLY inhibitor 326E as a promising treatment for hypercholesterolemia
- PMID: 36873173
- PMCID: PMC9979192
- DOI: 10.1016/j.apsb.2022.06.011
Development of the novel ACLY inhibitor 326E as a promising treatment for hypercholesterolemia
Abstract
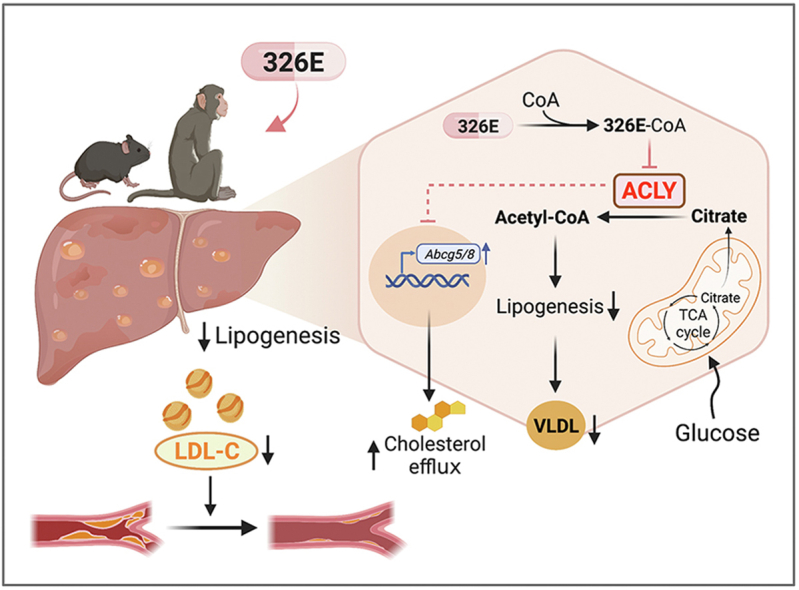
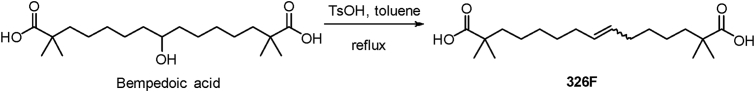
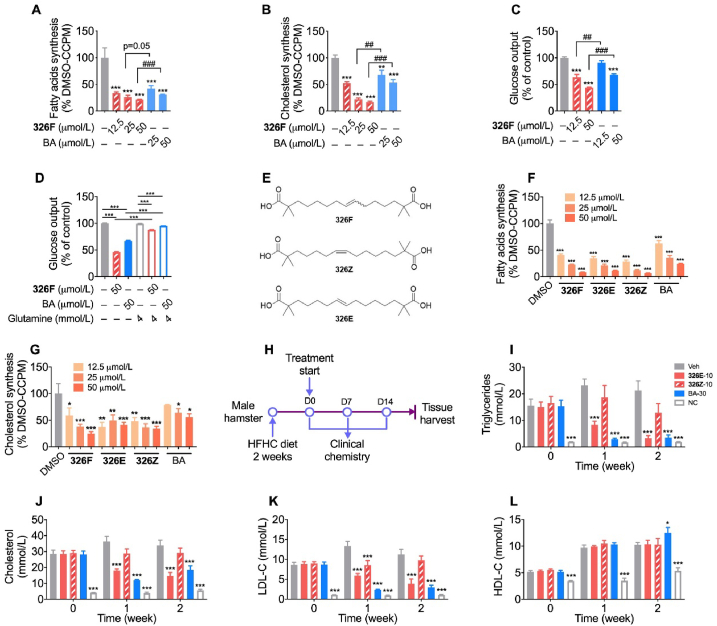
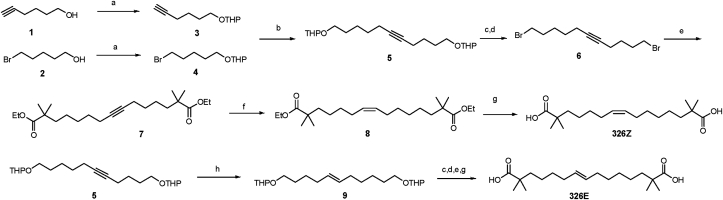
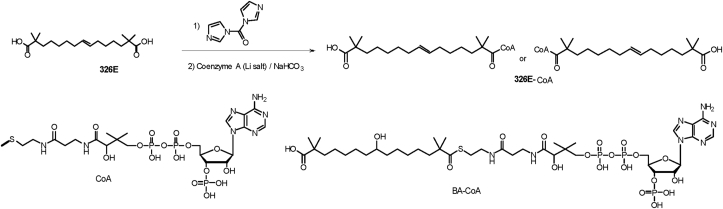
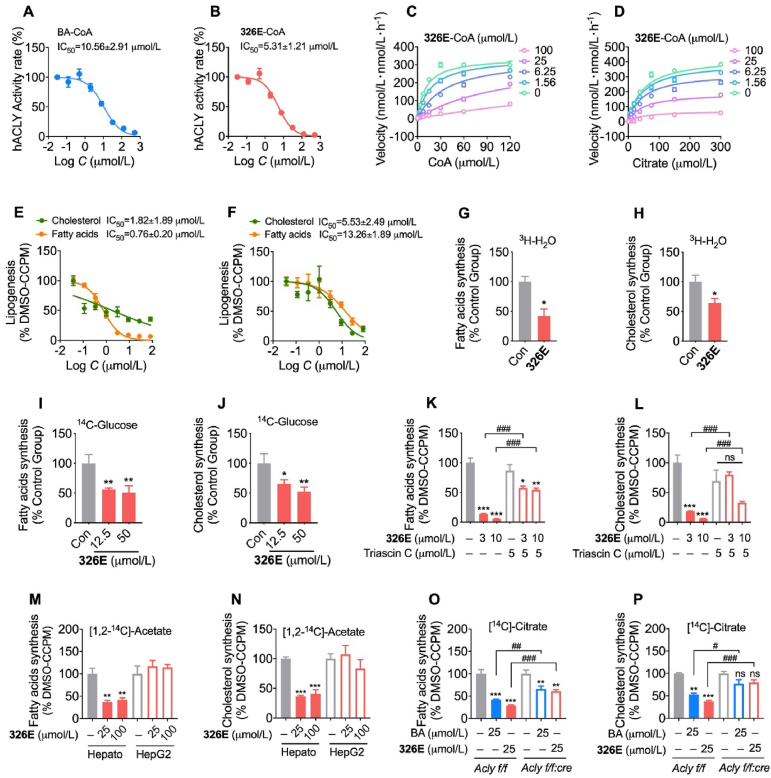
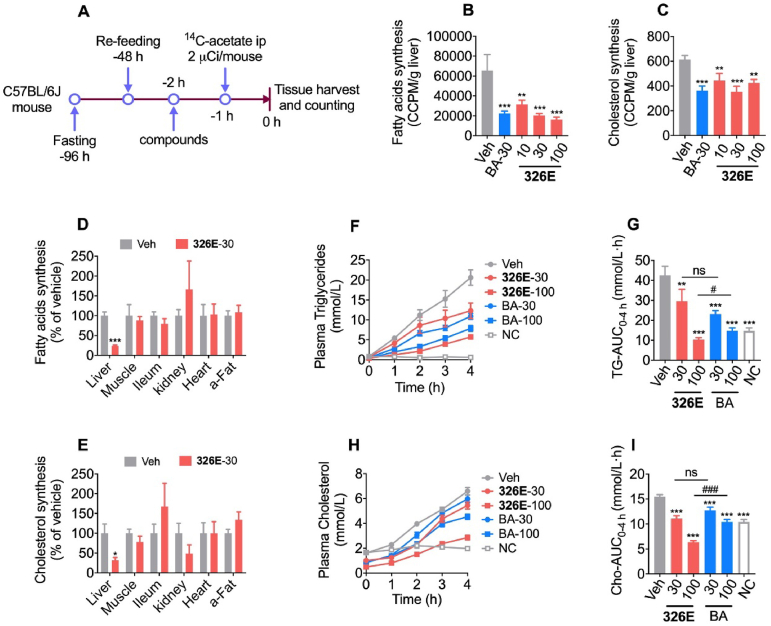
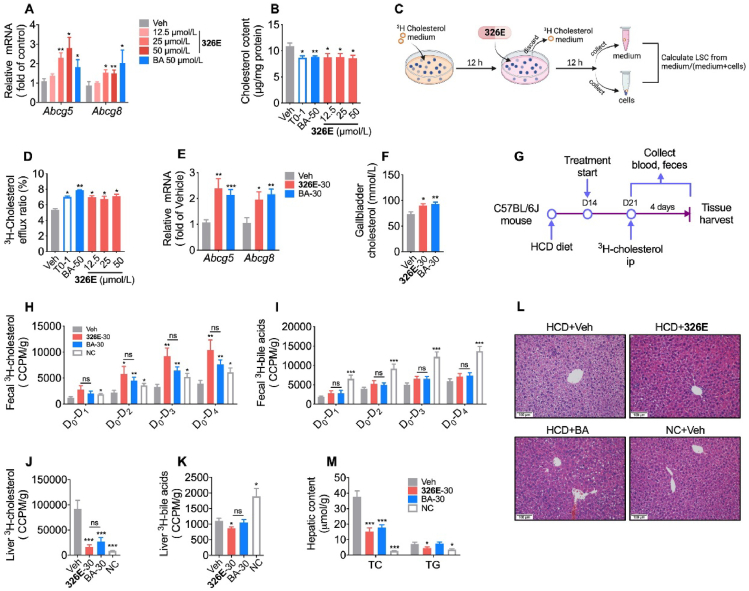
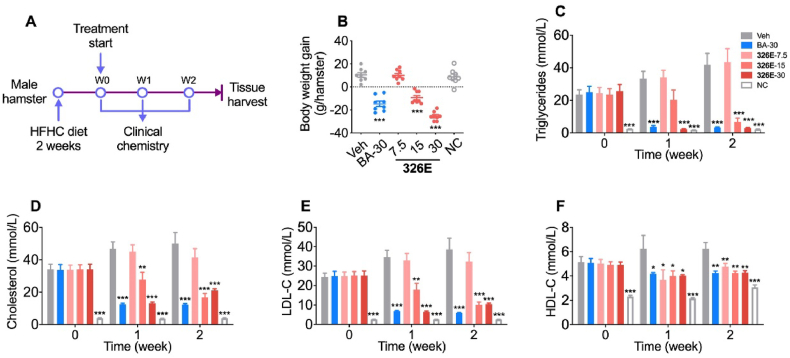
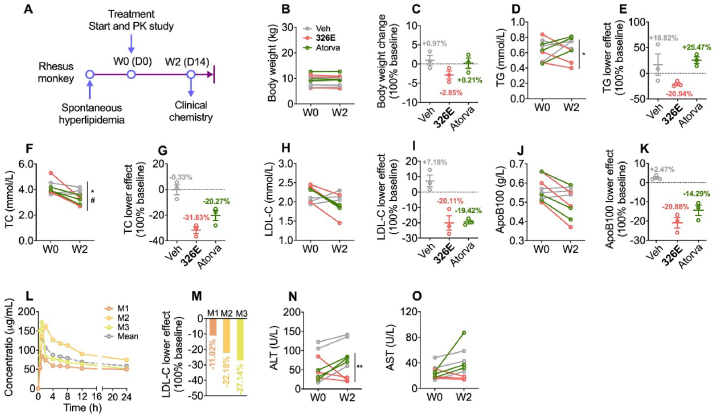
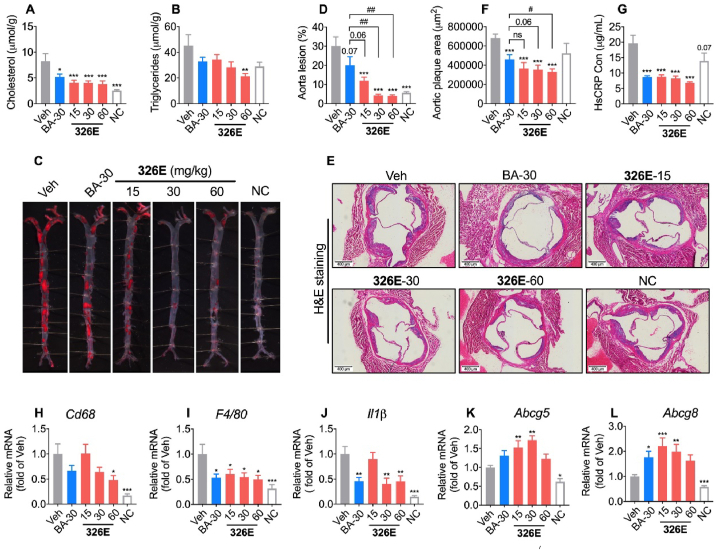
Hepatic cholesterol accumulation is an important contributor to hypercholesterolemia, which results in atherosclerosis and cardiovascular disease (CVD). ATP-citrate lyase (ACLY) is a key lipogenic enzyme that converts cytosolic citrate derived from tricarboxylic acid cycle (TCA cycle) to acetyl-CoA in the cytoplasm. Therefore, ACLY represents a link between mitochondria oxidative phosphorylation and cytosolic de novo lipogenesis. In this study, we developed the small molecule 326E with an enedioic acid structural moiety as a novel ACLY inhibitor, and its CoA-conjugated form 326E-CoA inhibited ACLY activity with an IC50 = 5.31 ± 1.2 μmol/L in vitro. 326E treatment reduced de novo lipogenesis, and increased cholesterol efflux in vitro and in vivo. 326E was rapidly absorbed after oral administration, exhibited a higher blood exposure than that of the approved ACLY inhibitor bempedoic acid (BA) used for hypercholesterolemia. Chronic 326E treatment in hamsters and rhesus monkeys resulted in remarkable improvement of hyperlipidemia. Once daily oral administration of 326E for 24 weeks prevented the occurrence of atherosclerosis in ApoE-/- mice to a greater extent than that of BA treatment. Taken together, our data suggest that inhibition of ACLY by 326E represents a promising strategy for the treatment of hypercholesterolemia.
Keywords: ACLY inhibitor; ATP-Citrate lyase (ACLY); Atherosclerosis; Cholesterol efflux; Hypercholesterolemia; Lipogenesis; Liver.
© 2022 Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences. Production and hosting by Elsevier B.V.
Conflict of interest statement
Yangming Zhang received consultant allowance from Burgeon Therapeutics Co., Ltd. The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Ference B.A., Ginsberg H.N., Graham I., Ray K.K., Packard C.J., Bruckert E., et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the european atherosclerosis society consensus panel. Eur Heart J. 2017;38:2459–2472. - PMC - PubMed
-
- Silverman M.G., Ference B.A., Im K., Wiviott S.D., Giugliano R.P., Grundy S.M., et al. Association between lowering LDL-C and cardiovascular risk reduction among different therapeutic interventions: a systematic review and meta-analysis. JAMA. 2016;316:1289–1297. - PubMed
-
- Stone N.J., Robinson J.G., Lichtenstein A.H., Bairey Merz C.N., Blum C.B., Eckel R.H., et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the american college of cardiology/american heart association task force on practice guidelines. Circulation. 2014;129:S1–S45. - PubMed
-
- Karr S. Epidemiology and management of hyperlipidemia. Am J Manag Care. 2017;23:S139–S148. - PubMed
-
- Xiao C., Dash S., Morgantini C., Hegele R.A., Lewis G.F. Pharmacological targeting of the atherogenic dyslipidemia complex: the next frontier in CVD prevention beyond lowering LDL cholesterol. Diabetes. 2016;65:1767–1778. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
