

Steroid-resistant nephrotic syndrome associated with certain SGPL1 variants in a family: Case report and literature review
- PMID: 36873630
- PMCID: PMC9978203
- DOI: 10.3389/fped.2023.1079758
Steroid-resistant nephrotic syndrome associated with certain SGPL1 variants in a family: Case report and literature review
Abstract
Objectives: Steroid-resistant nephrotic syndrome (SRNS) is a clinical syndrome characterized by the lack of response to standard steroid therapy, usually progressing to end-stage renal disease. We reported two cases of female identical twins with SRNS caused by SGPL1 variants in one family, reviewed the relevant literature, and summarized their clinical phenotypes, pathological types, and genotypic characteristics.
Methods: Two cases of nephrotic syndrome caused by SGPL1 variants were admitted to Tongji Hospital, affiliated with Tongji Medical College of Huazhong University of Science and Technology. Their clinical data were retrospectively collected, and the peripheral blood genomic DNA was captured and sequenced by whole exome sequencing. Related literature published in PubMed, CNKI, and Wan fang databases was reviewed.
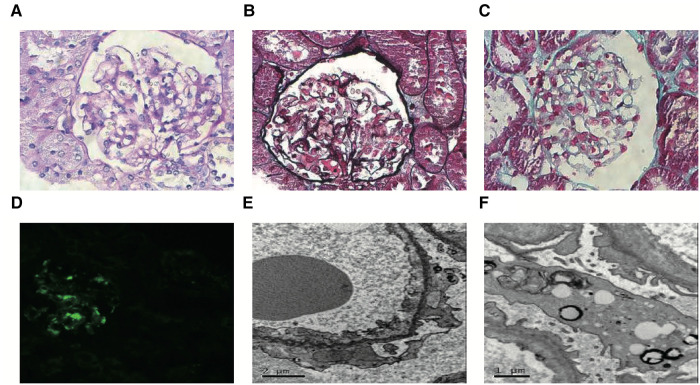
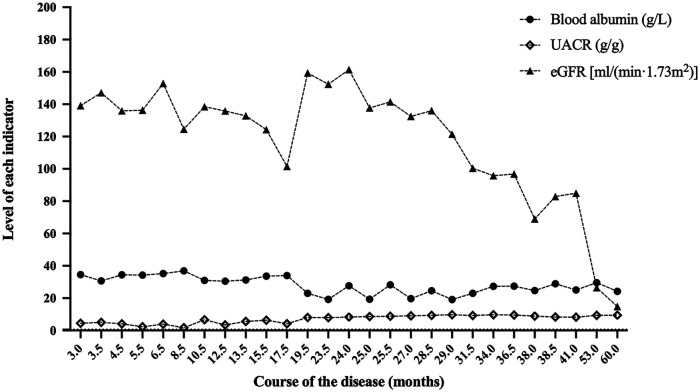
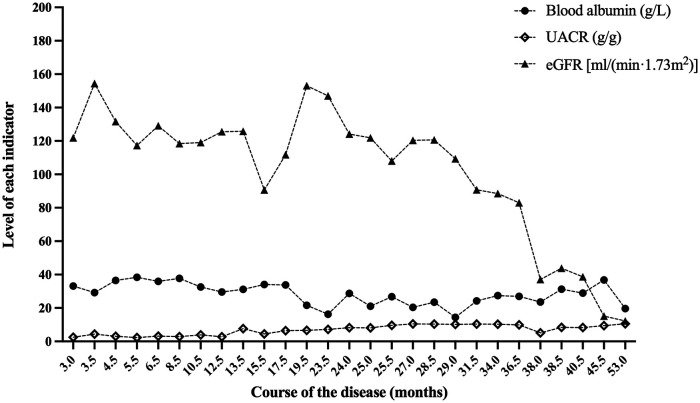
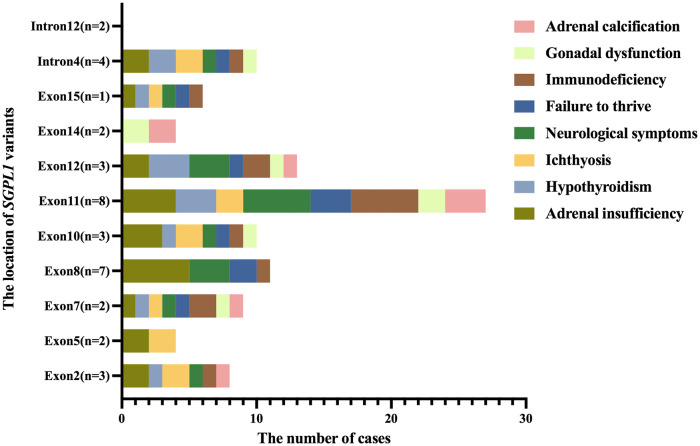
Results: We described two Chinese identical twin girls with isolated SRNS due to compound heterozygous variants in the SGPL1 (intron4 c.261 + 1G > A and intron12 c.1298 + 6T > C). The patients were followed up for 60.0 months and 53.0 months, respectively, having no extra-renal manifestations. They all died due to renal failure. A total of 31 children with SGPL1 variants causing nephrotic syndrome (including the reported two cases) were identified through a literature review.
Conclusions: These two female identical twins were the first reported cases of isolated SRNS caused by SGPL1 variants. Almost all homozygous and compound heterozygous variants of SGPL1 had extra-renal manifestations, but compound heterozygous variants in the intron of SGPL1 may have no obvious extra-renal manifestations. Additionally, a negative genetic testing result does not completely rule out genetic SRNS because the Human Gene Mutation Database or ClinVar is constantly being updated.
Keywords: SGPL1 gene mutation; clinical phenotype; genotype; proteinuria; steroid-resistant nephrotic syndrome.
© 2023 Yang, He, Zhou, Yuan and Qiu.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
LinkOut - more resources
Full Text Sources