

Diagnosis of an intermediate case of maple syrup urine disease: A case report
- PMID: 36874425
- PMCID: PMC9979284
- DOI: 10.12998/wjcc.v11.i5.1077
Diagnosis of an intermediate case of maple syrup urine disease: A case report
Abstract
Background: Maple syrup urine disease (MSUD) is an autosomal recessive genetic disorder caused by defects in the catabolism of the branched-chain amino acids (BCAAs). However, the clinical and metabolic screening is limited in identifying all MSUD patients, especially those patients with mild phenotypes or are asymptomatic. This study aims to share the diagnostic experience of an intermediate MSUD case who was missed by metabolic profiling but identified by genetic analysis.
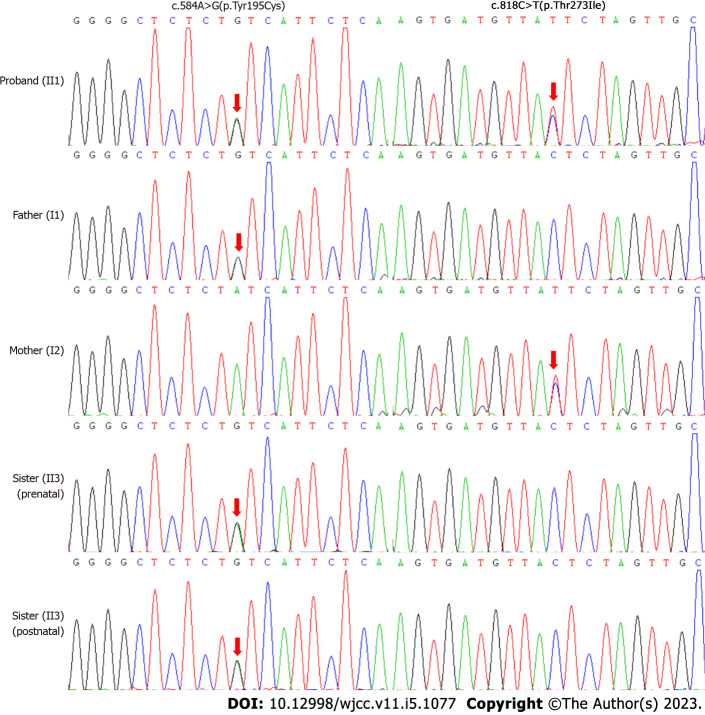
Case summary: This study reports the diagnostic process of a boy with intermediate MSUD. The proband presented with psychomotor retardation and cerebral lesions on magnetic resonance imaging scans at 8 mo of age. Preliminary clinical and metabolic profiling did not support a specific disease. However, whole exome sequencing and subsequent Sanger sequencing at 1 year and 7 mo of age identified bi-allelic pathogenic variants of the BCKDHB gene, confirming the proband as having MSUD with non-classic mild phenotypes. His clinical and laboratory data were retrospectively analyzed. According to his disease course, he was classified into an intermediate form of MSUD. His management was then changed to BCAAs restriction and metabolic monitoring conforming to MSUD. In addition, genetic counseling and prenatal diagnosis were provided to his parents.
Conclusion: Our work provides diagnostic experience of an intermediate MSUD case, suggesting that a genetic analysis is important for ambiguous cases, and alerts clinicians to avoid missing patients with non-classic mild phenotypes of MSUD.
Keywords: BCKDHB gene; Branched-chain amino acids; Case report; Genetic analysis; Maple syrup urine disease; Metabolic profiling.
©The Author(s) 2023. Published by Baishideng Publishing Group Inc. All rights reserved.
Conflict of interest statement
Conflict-of-interest statement: The authors declare that they have no conflict of interest.
Figures


References
-
- Chuang JL, Fisher CR, Cox RP, Chuang DT. Molecular basis of maple syrup urine disease: novel mutations at the E1 alpha locus that impair E1(alpha 2 beta 2) assembly or decrease steady-state E1 alpha mRNA levels of branched-chain alpha-keto acid dehydrogenase complex. Am J Hum Genet . 1994;55:297–304. - PMC - PubMed
-
- Fisher CW, Fisher CR, Chuang JL, Lau KS, Chuang DT, Cox RP. Occurrence of a 2-bp (AT) deletion allele and a nonsense (G-to-T) mutant allele at the E2 (DBT) locus of six patients with maple syrup urine disease: multiple-exon skipping as a secondary effect of the mutations. Am J Hum Genet . 1993;52:414–424. - PMC - PubMed
-
- Odièvre MH, Chretien D, Munnich A, Robinson BH, Dumoulin R, Masmoudi S, Kadhom N, Rötig A, Rustin P, Bonnefont JP. A novel mutation in the dihydrolipoamide dehydrogenase E3 subunit gene (DLD) resulting in an atypical form of alpha-ketoglutarate dehydrogenase deficiency. Hum Mutat. 2005;25:323–324. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
