

resVAE ensemble: Unsupervised identification of gene sets in multi-modal single-cell sequencing data using deep ensembles
- PMID: 36875765
- PMCID: PMC9975353
- DOI: 10.3389/fcell.2023.1091047
resVAE ensemble: Unsupervised identification of gene sets in multi-modal single-cell sequencing data using deep ensembles
Abstract
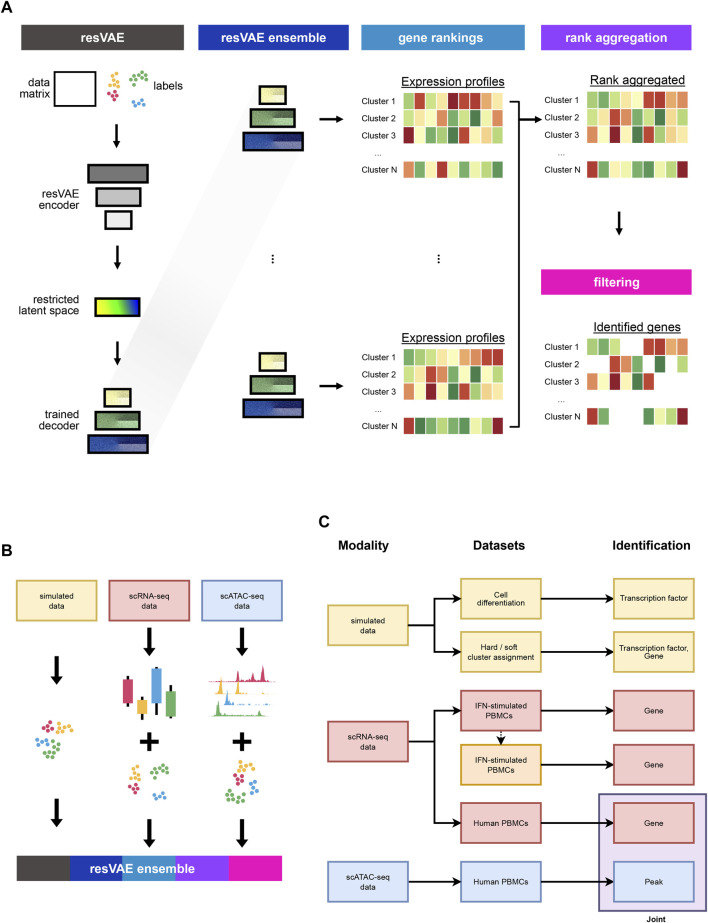
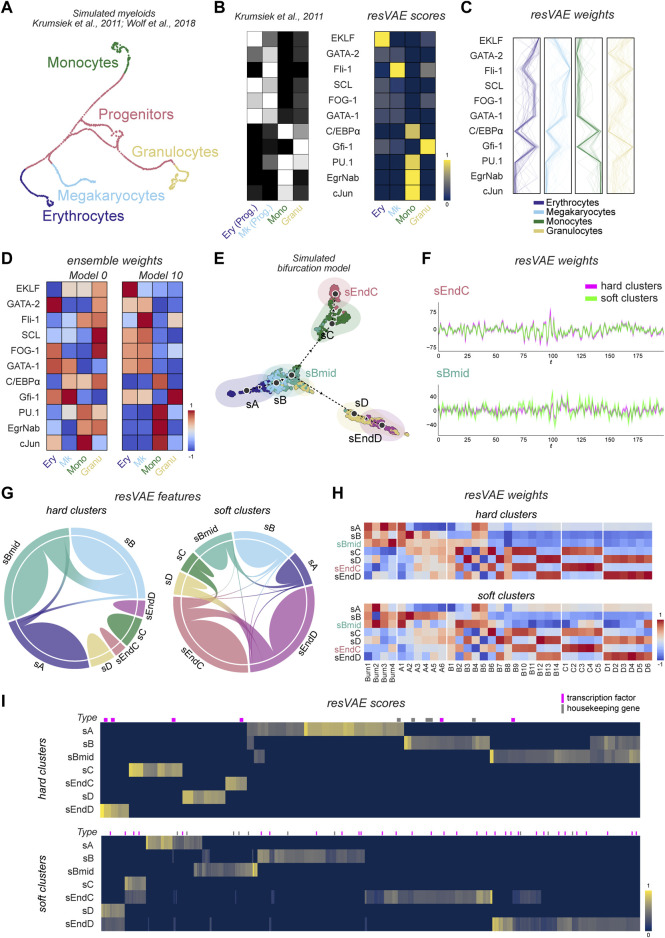
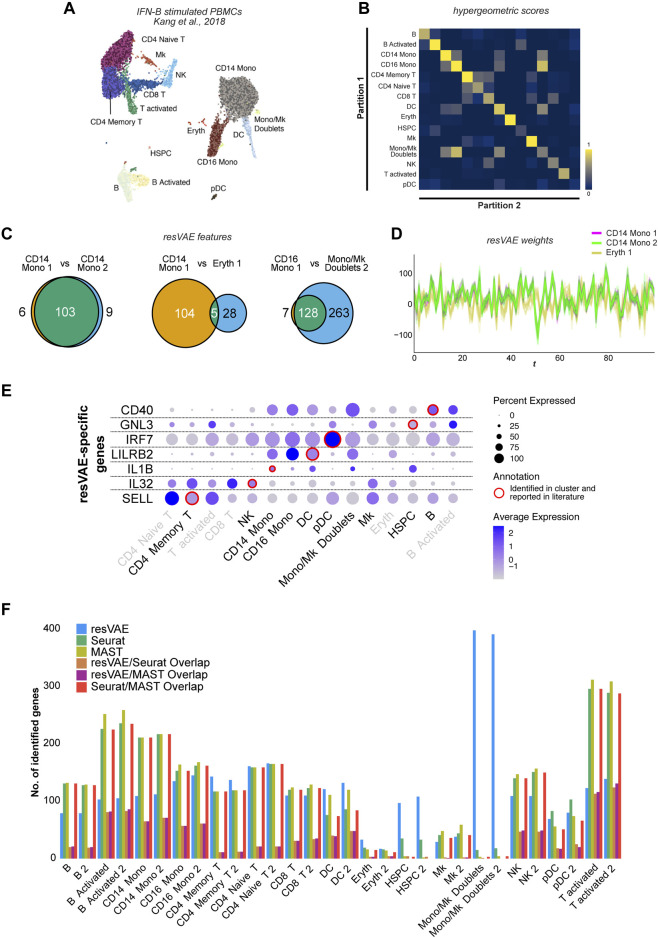
Feature identification and manual inspection is currently still an integral part of biological data analysis in single-cell sequencing. Features such as expressed genes and open chromatin status are selectively studied in specific contexts, cell states or experimental conditions. While conventional analysis methods construct a relatively static view on gene candidates, artificial neural networks have been used to model their interactions after hierarchical gene regulatory networks. However, it is challenging to identify consistent features in this modeling process due to the inherently stochastic nature of these methods. Therefore, we propose using ensembles of autoencoders and subsequent rank aggregation to extract consensus features in a less biased manner. Here, we performed sequencing data analyses of different modalities either independently or simultaneously as well as with other analysis tools. Our resVAE ensemble method can successfully complement and find additional unbiased biological insights with minimal data processing or feature selection steps while giving a measurement of confidence, especially for models using stochastic or approximation algorithms. In addition, our method can also work with overlapping clustering identity assignment suitable for transitionary cell types or cell fates in comparison to most conventional tools.
Keywords: bioinformatics; deep learning; ensemble; gene set analysis; rank aggregation; single-cell sequencing.
Copyright © 2023 Ten, Yuan, Jabareen, Phua, Eils, Lukassen and Conrad.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

References
-
- Datta L. (2020). A survey on activation functions and their relation with xavier and he normal initialization. arXiv preprint arXiv:2004.06632 .
LinkOut - more resources
Full Text Sources