

Microfluidics-free single-cell genomics with templated emulsification
- PMID: 36879006
- PMCID: PMC10635830
- DOI: 10.1038/s41587-023-01685-z
Microfluidics-free single-cell genomics with templated emulsification
Abstract
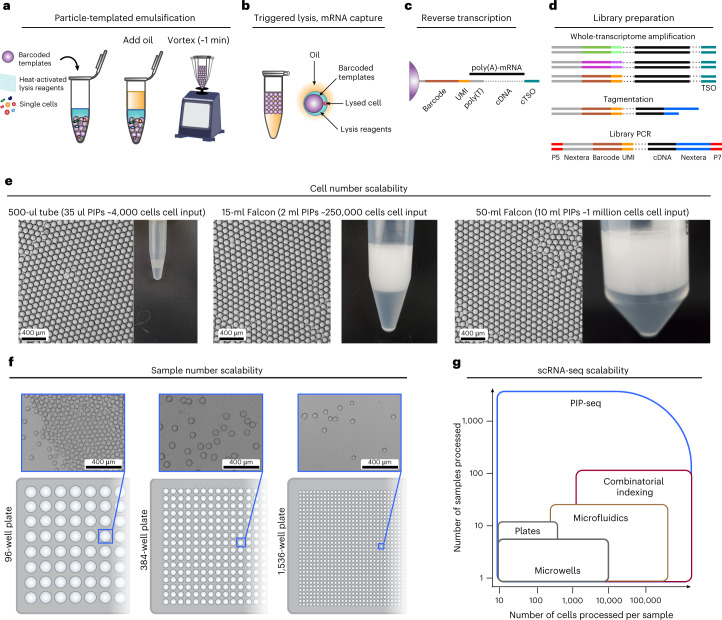
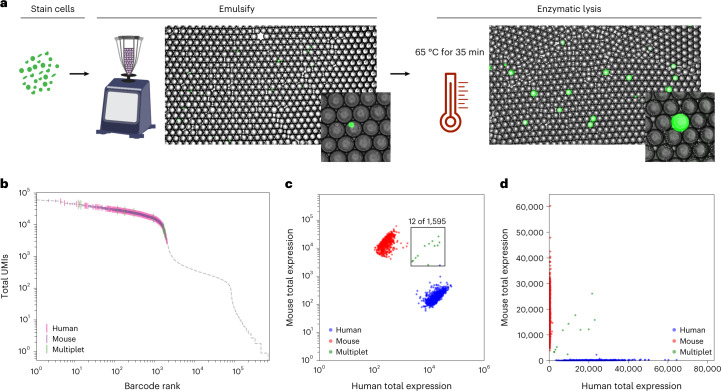
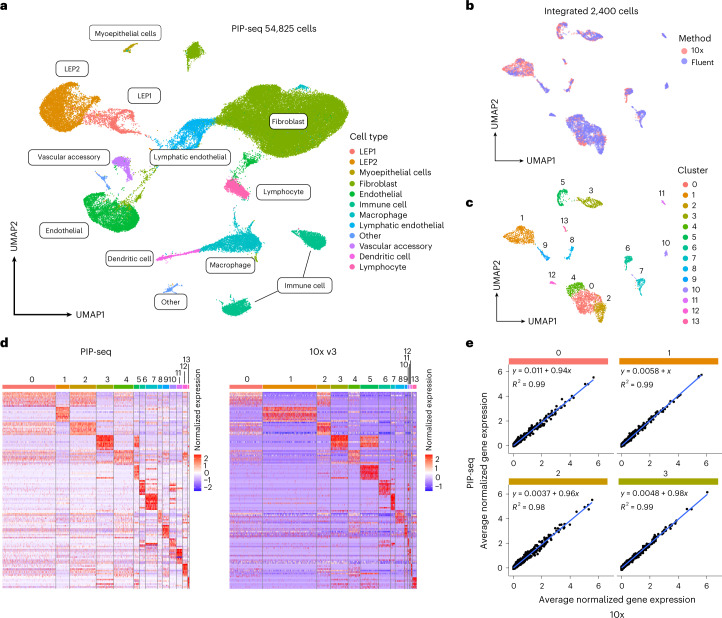
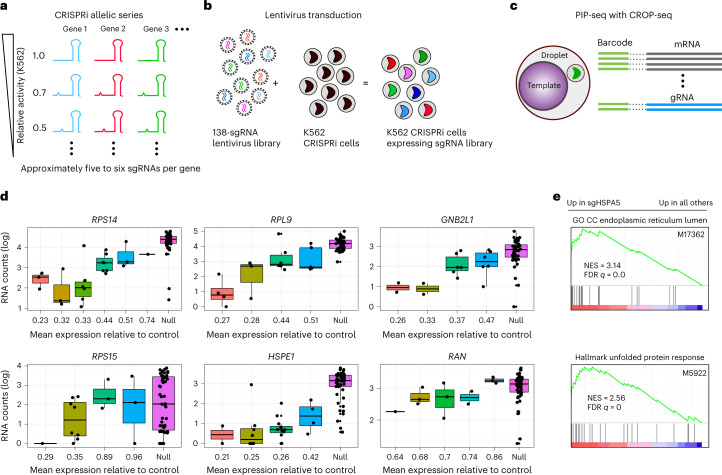
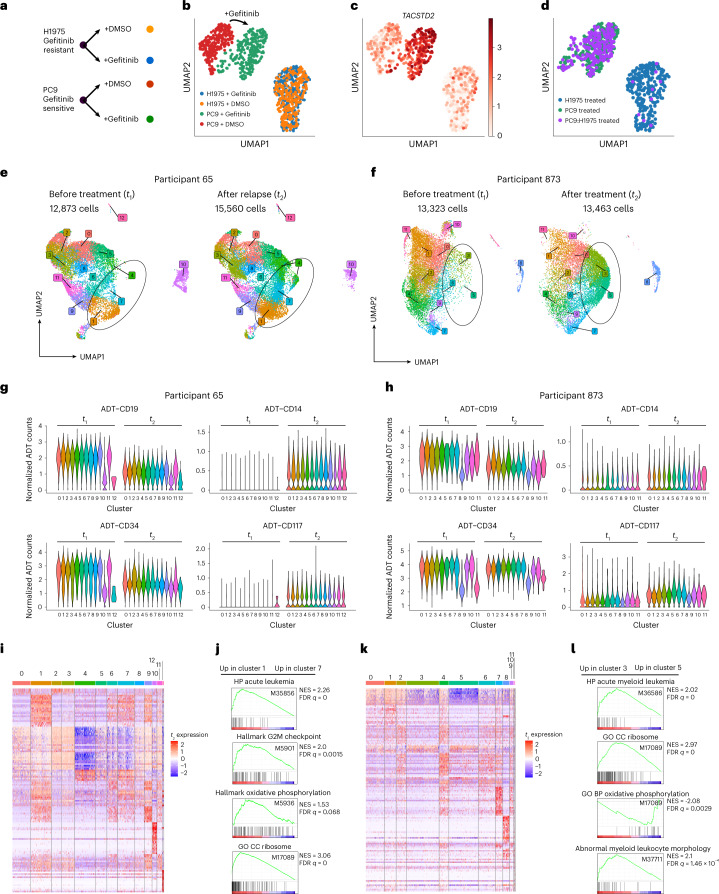
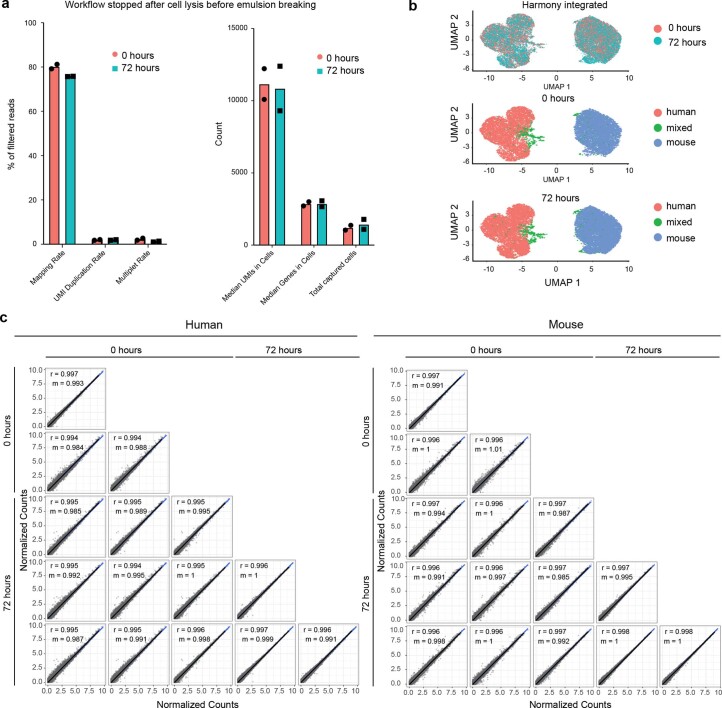
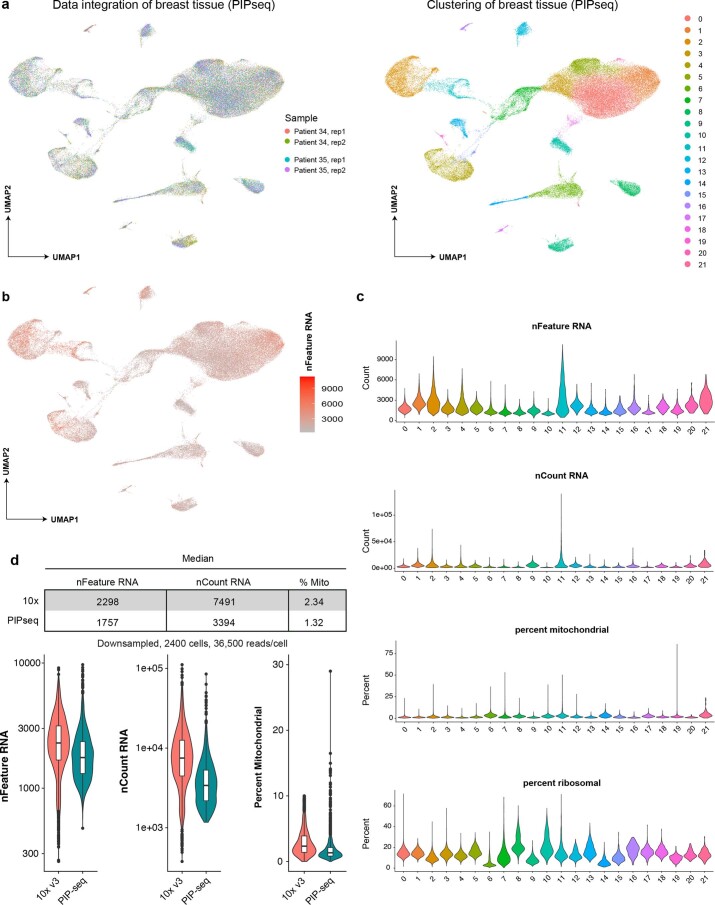
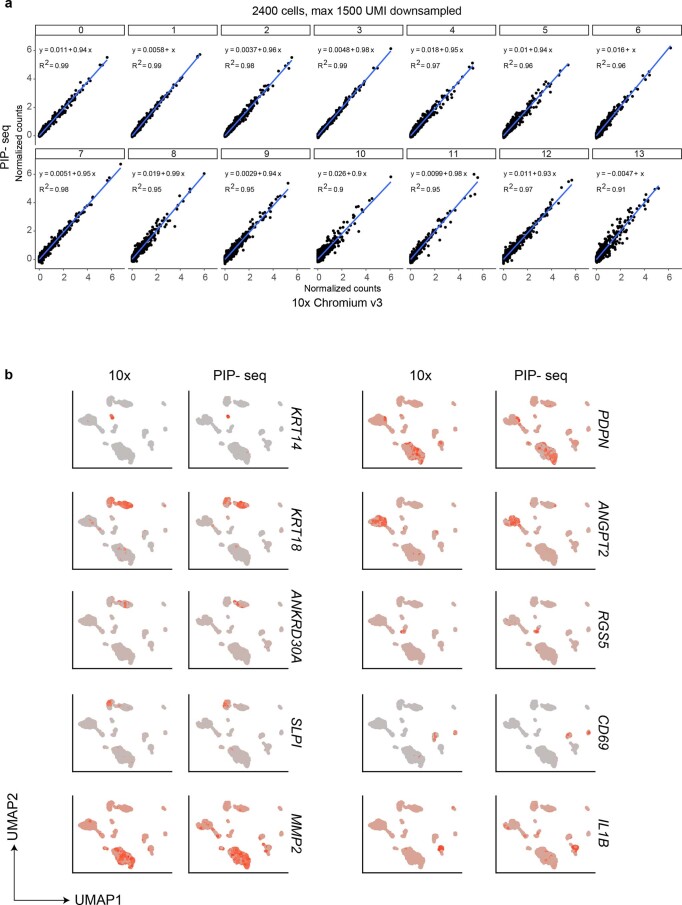
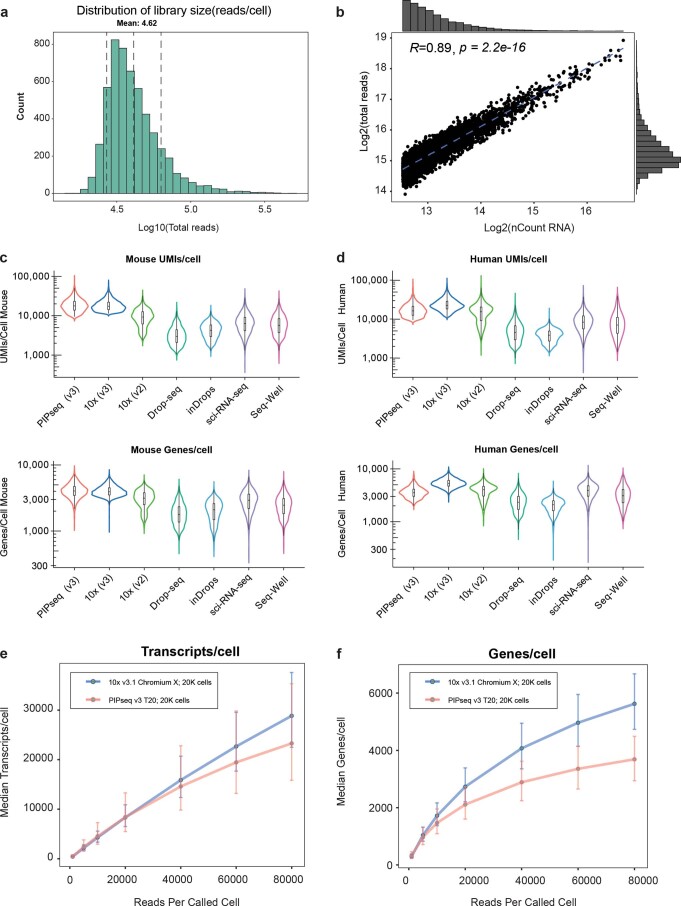
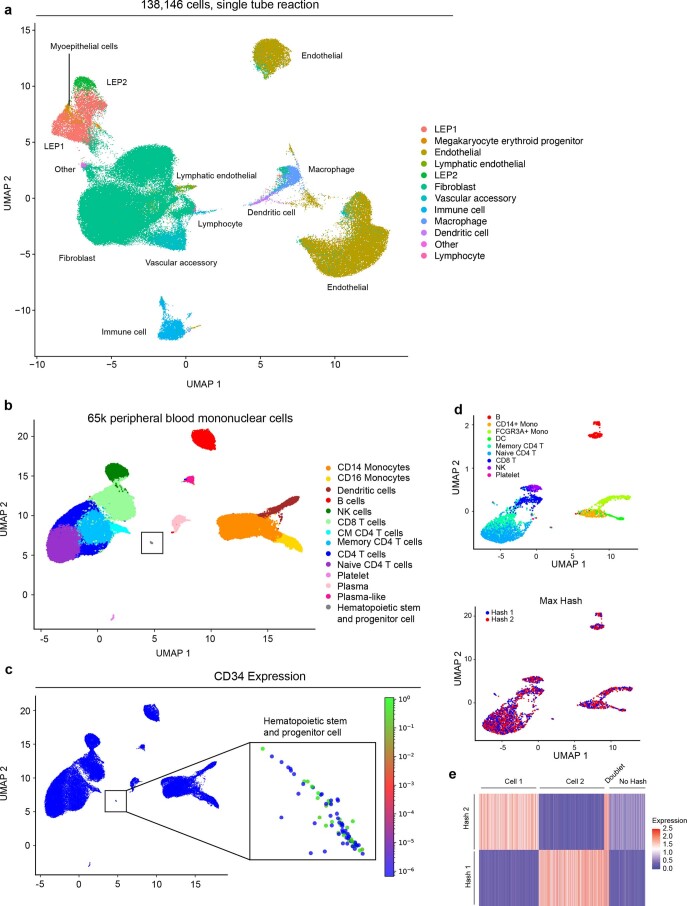
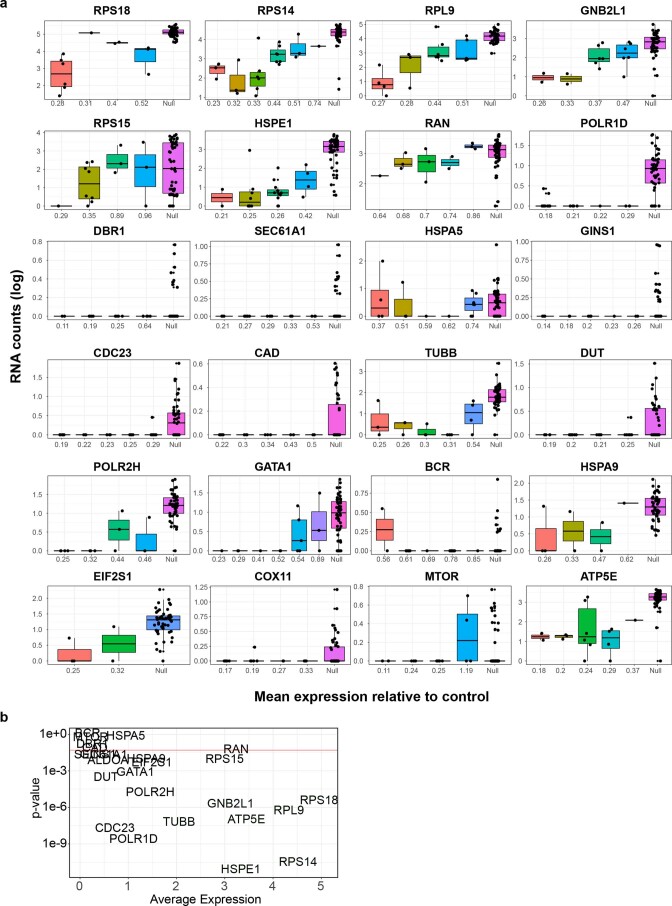
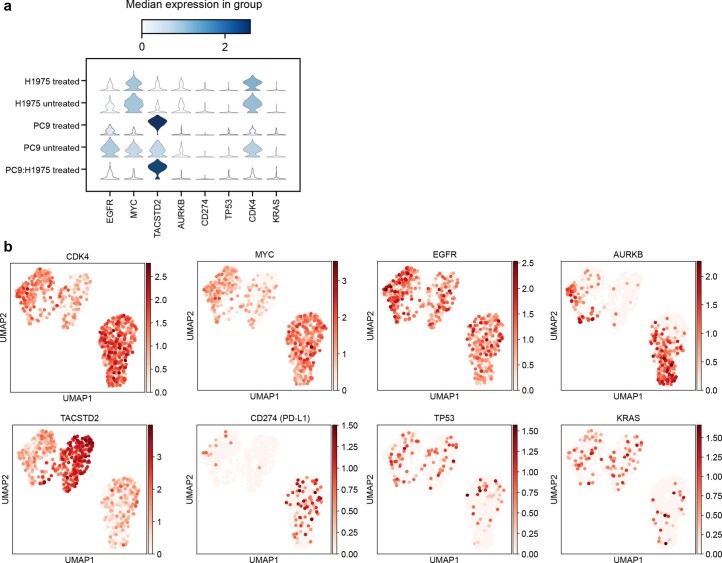
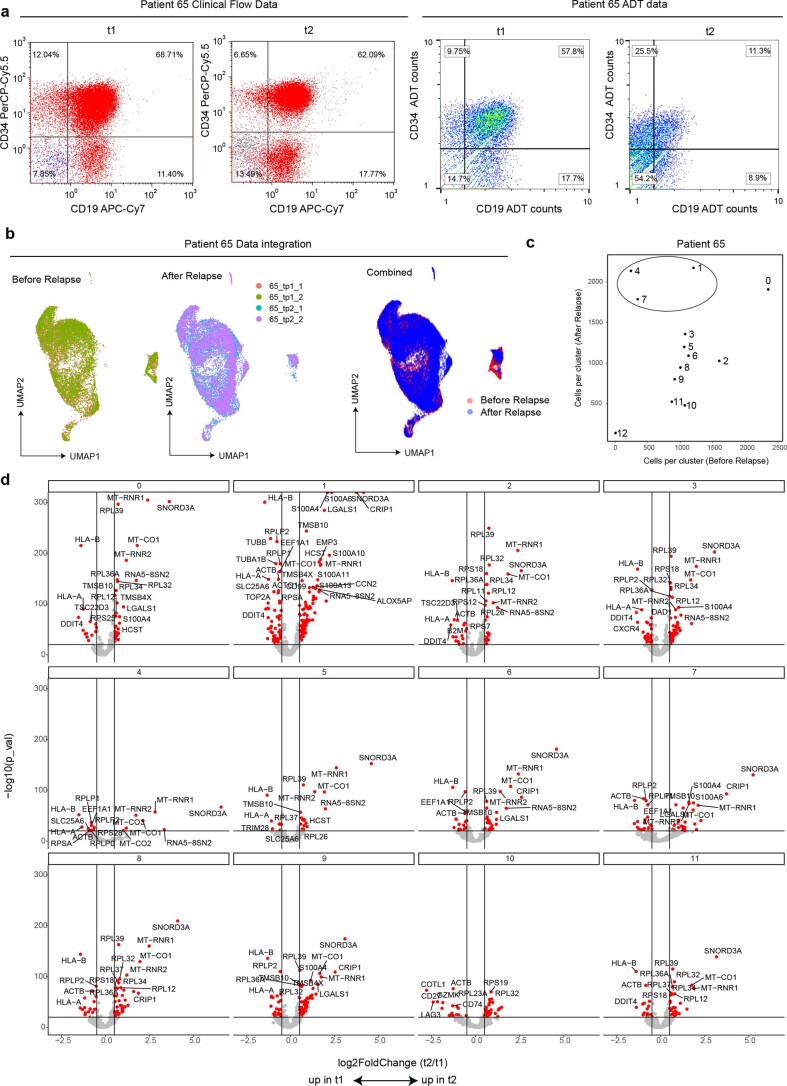
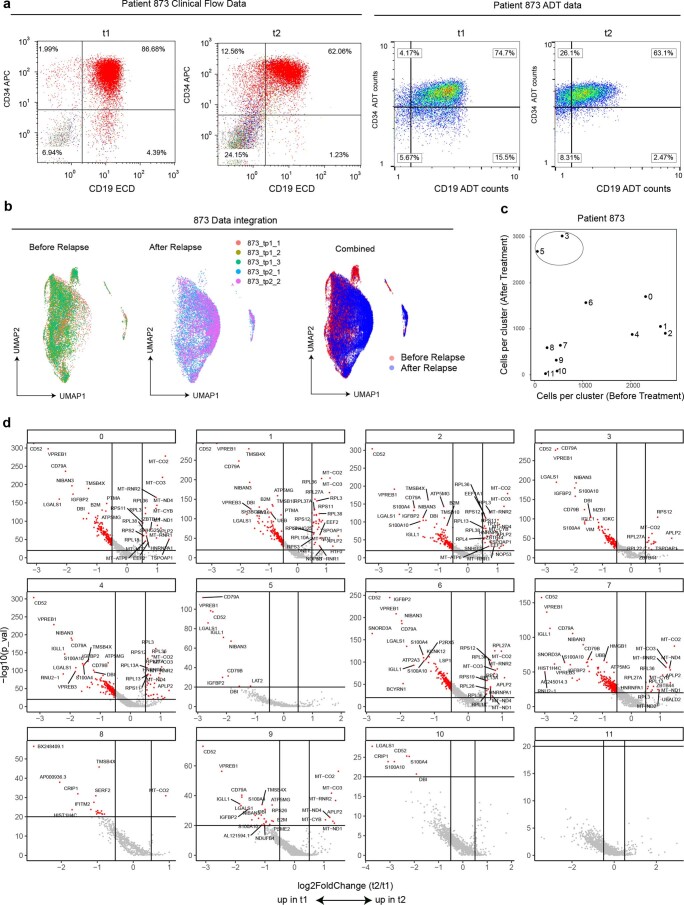
Current single-cell RNA-sequencing approaches have limitations that stem from the microfluidic devices or fluid handling steps required for sample processing. We develop a method that does not require specialized microfluidic devices, expertise or hardware. Our approach is based on particle-templated emulsification, which allows single-cell encapsulation and barcoding of cDNA in uniform droplet emulsions with only a vortexer. Particle-templated instant partition sequencing (PIP-seq) accommodates a wide range of emulsification formats, including microwell plates and large-volume conical tubes, enabling thousands of samples or millions of cells to be processed in minutes. We demonstrate that PIP-seq produces high-purity transcriptomes in mouse-human mixing studies, is compatible with multiomics measurements and can accurately characterize cell types in human breast tissue compared to a commercial microfluidic platform. Single-cell transcriptional profiling of mixed phenotype acute leukemia using PIP-seq reveals the emergence of heterogeneity within chemotherapy-resistant cell subsets that were hidden by standard immunophenotyping. PIP-seq is a simple, flexible and scalable next-generation workflow that extends single-cell sequencing to new applications.
© 2023. The Author(s).
Conflict of interest statement
A.R.A. filed a patent related to templated emulsification and is a founder of Fluent Biosciences. I.C.C. consults for Fluent Biosciences and is on its Scientific Advisory Board. K.M.F., R.H.M., Y.X., C.H., A.O., P.H., J.S.A.I., J.Q.Z., A.M.-Z. and C.D.A. are employees at Fluent Biosciences and are working to commercialize the PIP-seq technology. M.J. consults for Maze Therapeutics and Gate Biosciences. J.M.R. consults for Maze Therapeutics and Waypoint Bio. J.S.W. declares outside interest in 5 AM Venture, Amgen, Chroma Medicine, DEM Biosciences, KSQ Therapeutics, Maze Therapeutics, Tenaya Therapeutics, Tessera Therapeutics and Velia Therapeutics. All other authors have no competing interests to declare.
Figures

References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
