

Characterization of neurotropic HPAI H5N1 viruses with novel genome constellations and mammalian adaptive mutations in free-living mesocarnivores in Canada
- PMID: 36880345
- PMCID: PMC10026807
- DOI: 10.1080/22221751.2023.2186608
Characterization of neurotropic HPAI H5N1 viruses with novel genome constellations and mammalian adaptive mutations in free-living mesocarnivores in Canada
Abstract
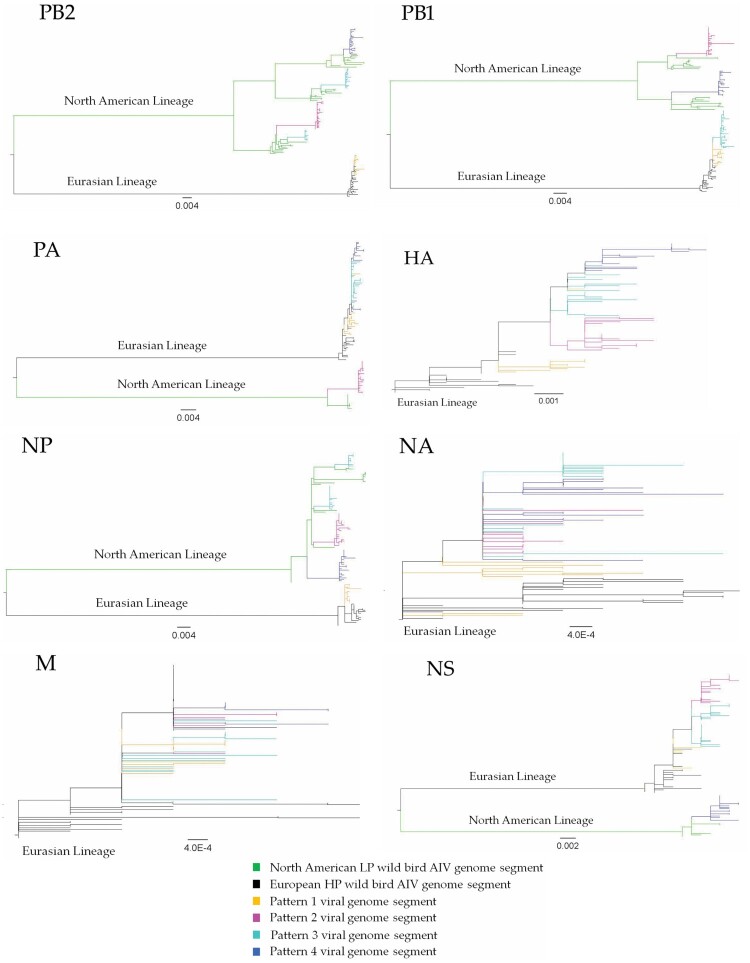
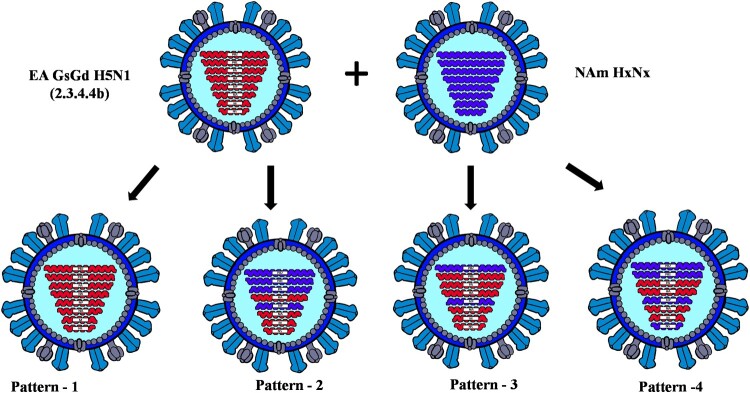
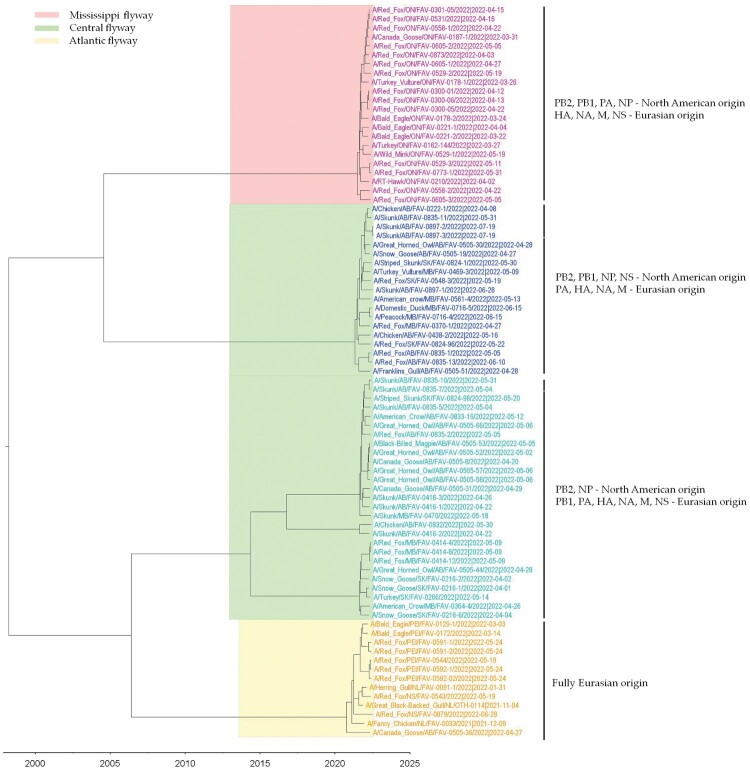
The GsGd lineage (A/goose/Guangdong/1/1996) H5N1 virus was introduced to Canada in 2021/2022 through the Atlantic and East Asia-Australasia/Pacific flyways by migratory birds. This was followed by unprecedented outbreaks affecting domestic and wild birds, with spillover into other animals. Here, we report sporadic cases of H5N1 in 40 free-living mesocarnivore species such as red foxes, striped skunks, and mink in Canada. The clinical presentations of the disease in mesocarnivores were consistent with central nervous system infection. This was supported by the presence of microscopic lesions and the presence of abundant IAV antigen by immunohistochemistry. Some red foxes that survived clinical infection developed anti-H5N1 antibodies. Phylogenetically, the H5N1 viruses from the mesocarnivore species belonged to clade 2.3.4.4b and had four different genome constellation patterns. The first group of viruses had wholly Eurasian (EA) genome segments. The other three groups were reassortant viruses containing genome segments derived from both North American (NAm) and EA influenza A viruses. Almost 17 percent of the H5N1 viruses had mammalian adaptive mutations (E627 K, E627V and D701N) in the polymerase basic protein 2 (PB2) subunit of the RNA polymerase complex. Other mutations that may favour adaptation to mammalian hosts were also present in other internal gene segments. The detection of these critical mutations in a large number of mammals within short duration after virus introduction inevitably highlights the need for continually monitoring and assessing mammalian-origin H5N1 clade 2.3.4.4b viruses for adaptive mutations, which potentially can facilitate virus replication, horizontal transmission and posing pandemic risks for humans.
Keywords: Clade 2.3.4.4b; H5N1; HPAI; mammals; mutation; reassortment.
Conflict of interest statement
No potential conflict of interest was reported by the author(s).
Figures
References
-
- Klenk HD, Rott R, Becht H.. On the structure of the influenza virus envelope. Virology. 1972;47:579–591. - PubMed
-
- Shaw ML, Palese P.. Orthomyxoviridae. In: Knipe D, Howley P, editor. Fields virology. Philadelphia, PA: Lippincott Williams & Wilkins; 2013. p. 1151–1185.
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical