

β3AR-Dependent Brain-Derived Neurotrophic Factor (BDNF) Generation Limits Chronic Postischemic Heart Failure
- PMID: 36884028
- PMCID: PMC10281793
- DOI: 10.1161/CIRCRESAHA.122.321583
β3AR-Dependent Brain-Derived Neurotrophic Factor (BDNF) Generation Limits Chronic Postischemic Heart Failure
Abstract
Background: Loss of brain-derived neurotrophic factor (BDNF)/TrkB (tropomyosin kinase receptor B) signaling accounts for brain and cardiac disorders. In neurons, β-adrenergic receptor stimulation enhances local BDNF expression. It is unclear if this occurs in a pathophysiological relevant manner in the heart, especially in the β-adrenergic receptor-desensitized postischemic myocardium. Nor is it fully understood whether and how TrkB agonists counter chronic postischemic left ventricle (LV) decompensation, a significant unmet clinical milestone.
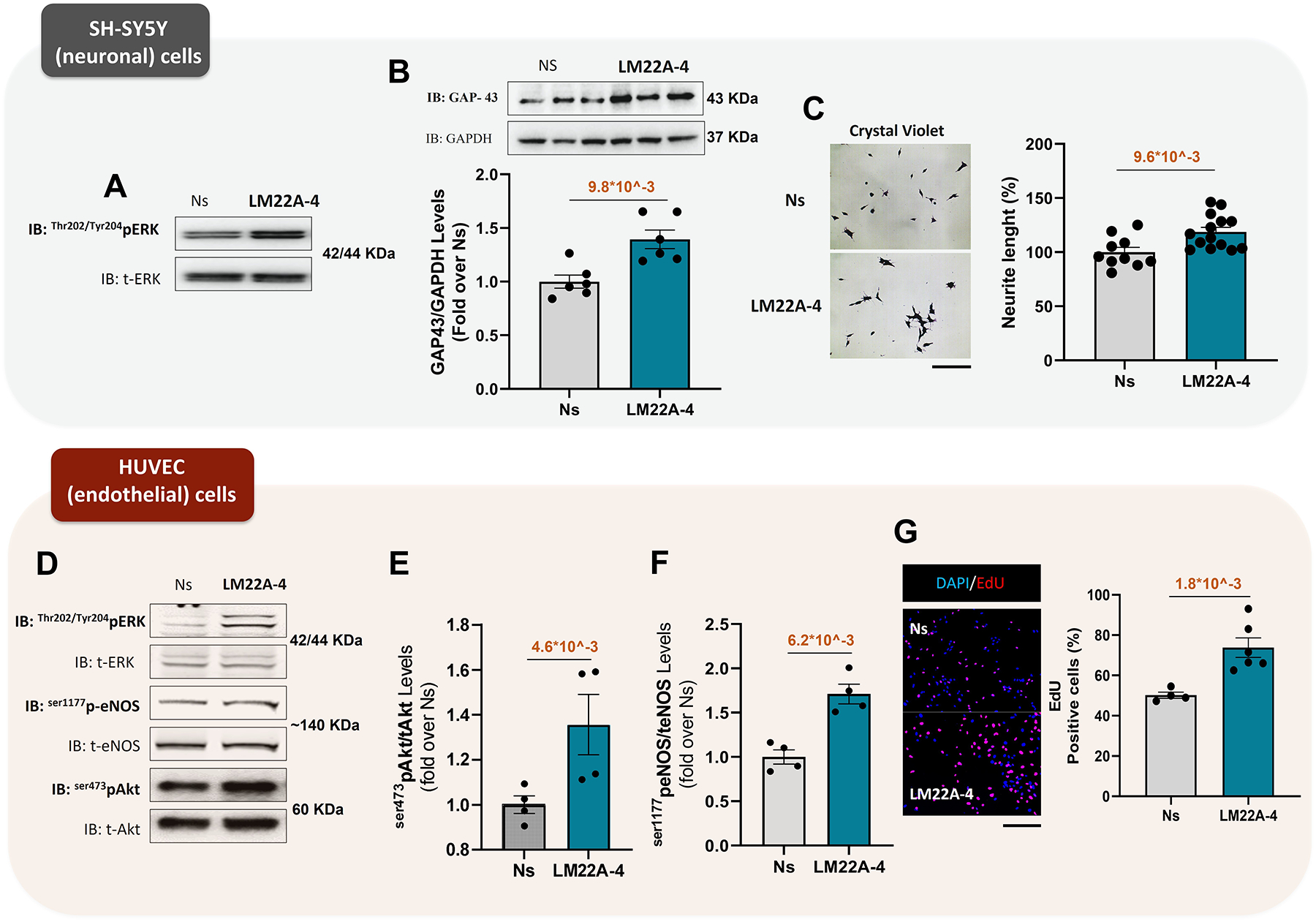
Methods: We conducted in vitro studies using neonatal rat and adult murine cardiomyocytes, SH-SY5Y neuronal cells, and umbilical vein endothelial cells. We assessed myocardial ischemia (MI) impact in wild type, β3AR knockout, or myocyte-selective BDNF knockout (myoBDNF KO) mice in vivo (via coronary ligation [MI]) or in isolated hearts with global ischemia-reperfusion (I/R).
Results: In wild type hearts, BDNF levels rose early after MI (<24 hours), plummeting at 4 weeks when LV dysfunction, adrenergic denervation, and impaired angiogenesis ensued. The TrkB agonist, LM22A-4, countered all these adverse effects. Compared with wild type, isolated myoBDNF KO hearts displayed worse infarct size/LV dysfunction after I/R injury and modest benefits from LM22A-4. In vitro, LM22A-4 promoted neurite outgrowth and neovascularization, boosting myocyte function, effects reproduced by 7,8-dihydroxyflavone, a chemically unrelated TrkB agonist. Superfusing myocytes with the β3AR-agonist, BRL-37344, increased myocyte BDNF content, while β3AR signaling underscored BDNF generation/protection in post-MI hearts. Accordingly, the β1AR blocker, metoprolol, via upregulated β3ARs, improved chronic post-MI LV dysfunction, enriching the myocardium with BDNF. Last, BRL-37344-imparted benefits were nearly abolished in isolated I/R injured myoBDNF KO hearts.
Conclusions: BDNF loss underscores chronic postischemic heart failure. TrkB agonists can improve ischemic LV dysfunction via replenished myocardial BDNF content. Direct cardiac β3AR stimulation, or β-blockers (via upregulated β3AR), is another BDNF-based means to fend off chronic postischemic heart failure.
Keywords: brain-derived neurotrophic factor; cardiac disorders; heart failure; myocardial ischemia; receptors, adrenergic.
Figures







References
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL157151/HL/NHLBI NIH HHS/United States
- P30 AG021334/AG/NIA NIH HHS/United States
- R01 HL061690/HL/NHLBI NIH HHS/United States
- R03 HL164393/HL/NHLBI NIH HHS/United States
- P01 HL134608/HL/NHLBI NIH HHS/United States
- R01 HL136918/HL/NHLBI NIH HHS/United States
- R01 HL063030/HL/NHLBI NIH HHS/United States
- P01 HL091799/HL/NHLBI NIH HHS/United States
- P01 HL075443/HL/NHLBI NIH HHS/United States
- P01 HL147841/HL/NHLBI NIH HHS/United States
- R37 HL061690/HL/NHLBI NIH HHS/United States
- R01 HL088503/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
