

Long-term Observation of a Japanese Patient with a Multiple-system Neurodegenerative Disorder with a Uniallelic de novo Missense Variant in KIF1A
- PMID: 36889712
- PMCID: PMC10641196
- DOI: 10.2169/internalmedicine.1184-22
Long-term Observation of a Japanese Patient with a Multiple-system Neurodegenerative Disorder with a Uniallelic de novo Missense Variant in KIF1A
Abstract
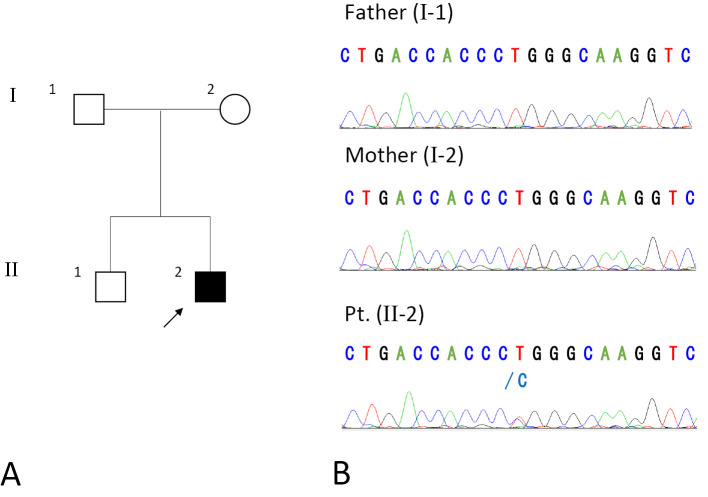
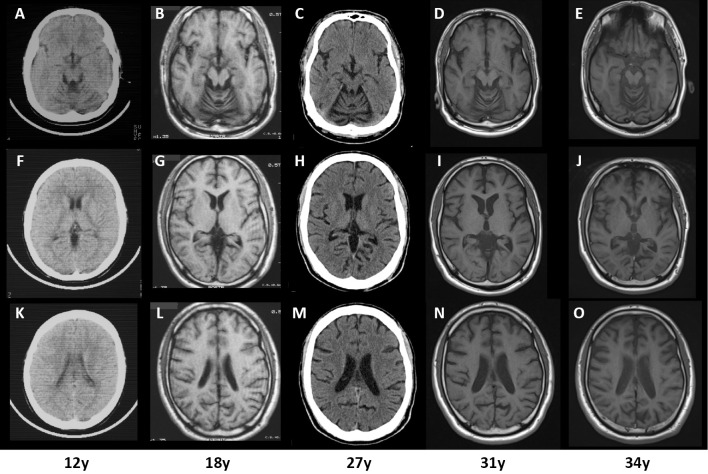
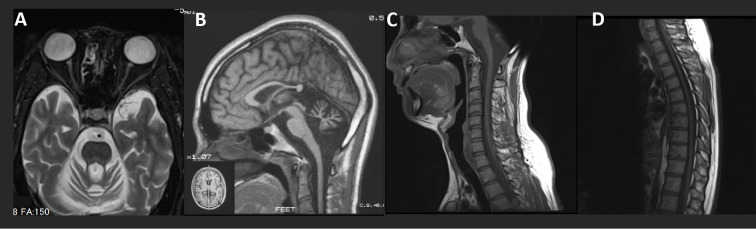
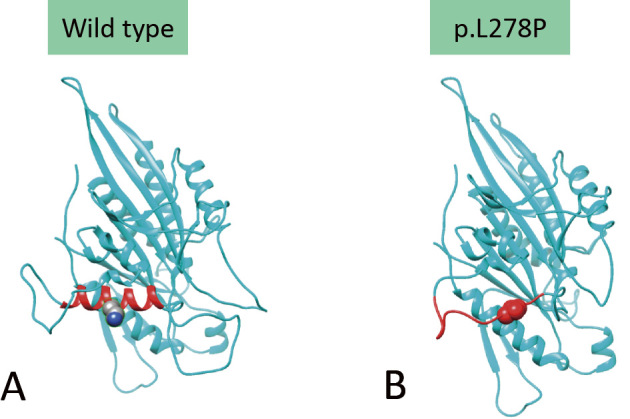
We encountered a 37-year-old Japanese man with KIF1A-associated neurological disorder (KAND) who exhibited motor developmental delay, intellectual disability, and slowly progressive cerebellar ataxia, hypotonia, and optic neuropathy. Pyramidal tract signs were evident late in this case. At 30 years old, the patient developed a neurogenic bladder. A molecular diagnosis revealed a uniallelic missense de novo variant (p.L278P) of KIF1A. Serial neuroradiological studies revealed atrophy of the cerebellum at an early age, and cerebral hemisphere atrophy progressed slowly over a 22-year observation period. Our study suggests that the primary etiology of KAND may be acquired, long-standing neurodegeneration rather than congenital hypoplasia.
Keywords: KAND; KIF1A; ataxia; intellectual disability; multiple-system neurodegeneration.
Conflict of interest statement
Figures

Similar articles
-
A Novel de novo KIF1A Mutation in a Patient with Ataxia, Intellectual Disability and Mild Foot Deformity.Cerebellum. 2023 Dec;22(6):1308-1311. doi: 10.1007/s12311-022-01489-y. Epub 2022 Oct 13. Cerebellum. 2023. PMID: 36227410 Free PMC article.
-
KIF1A mutation in a patient with progressive neurodegeneration.J Hum Genet. 2014 Nov;59(11):639-41. doi: 10.1038/jhg.2014.80. Epub 2014 Sep 25. J Hum Genet. 2014. PMID: 25253658
-
Novel KIF1A Variant in a Patient with Cerebellar Atrophy and Ataxia: A Case Report.Cerebellum. 2025 Apr 8;24(3):83. doi: 10.1007/s12311-025-01836-9. Cerebellum. 2025. PMID: 40198464
-
De novo dominant variants affecting the motor domain of KIF1A are a cause of PEHO syndrome.Eur J Hum Genet. 2016 Jun;24(6):949-53. doi: 10.1038/ejhg.2015.217. Epub 2015 Oct 21. Eur J Hum Genet. 2016. PMID: 26486474 Free PMC article. Review.
-
A novel pathogenic variant in the 3' end of the AGTPBP1 gene gives rise to neurodegeneration without cerebellar atrophy: an expansion of the disease phenotype?Neurogenetics. 2021 May;22(2):127-132. doi: 10.1007/s10048-021-00643-8. Epub 2021 Apr 28. Neurogenetics. 2021. PMID: 33909173 Review.
Cited by
-
Understanding speech and language in KIF1A-associated neurological disorder.Eur J Hum Genet. 2025 May 16. doi: 10.1038/s41431-025-01867-0. Online ahead of print. Eur J Hum Genet. 2025. PMID: 40379967
References
-
- Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol 10: 682-696, 2009. - PubMed
-
- Okada Y, Yamazaki H, Sekine-Aizawa Y, Hirokawa N. The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell 81: 769-780, 1995. - PubMed