

TaME-seq2: tagmentation-assisted multiplex PCR enrichment sequencing for viral genomic profiling
- PMID: 36890572
- PMCID: PMC9993372
- DOI: 10.1186/s12985-023-02002-5
TaME-seq2: tagmentation-assisted multiplex PCR enrichment sequencing for viral genomic profiling
Abstract
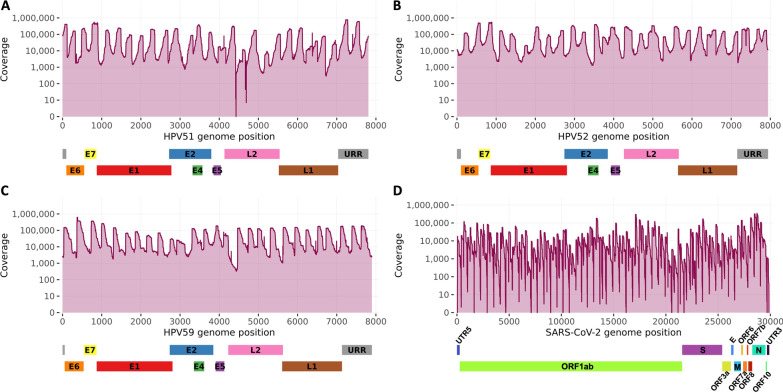
Background: Previously developed TaME-seq method for deep sequencing of HPV, allowed simultaneous identification of the human papillomavirus (HPV) DNA consensus sequence, low-frequency variable sites, and chromosomal integration events. The method has been successfully validated and applied to the study of five carcinogenic high-risk (HR) HPV types (HPV16, 18, 31, 33, and 45). Here, we present TaME-seq2 with an updated laboratory workflow and bioinformatics pipeline. The HR-HPV type repertoire was expanded with HPV51, 52, and 59. As a proof-of-concept, TaME-seq2 was applied on SARS-CoV-2 positive samples showing the method's flexibility to a broader range of viruses, both DNA and RNA.
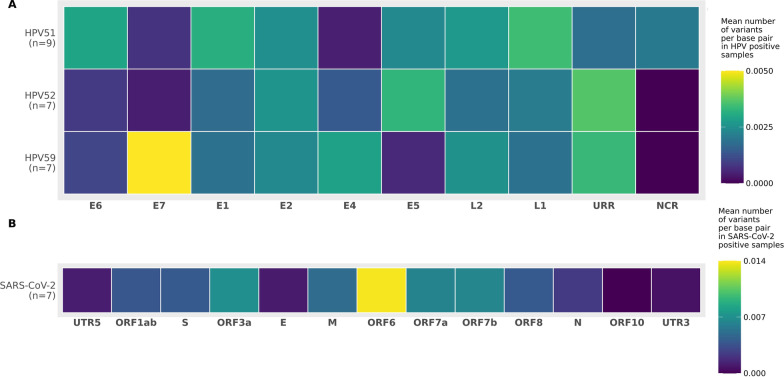
Results: Compared to TaME-seq version 1, the bioinformatics pipeline of TaME-seq2 is approximately 40× faster. In total, 23 HPV-positive samples and seven SARS-CoV-2 clinical samples passed the threshold of 300× mean depth and were submitted to further analysis. The mean number of variable sites per 1 kb was ~ 1.5× higher in SARS-CoV-2 than in HPV-positive samples. Reproducibility and repeatability of the method were tested on a subset of samples. A viral integration breakpoint followed by a partial genomic deletion was found in within-run replicates of HPV59-positive sample. Identified viral consensus sequence in two separate runs was > 99.9% identical between replicates, differing by a couple of nucleotides identified in only one of the replicates. Conversely, the number of identical minor nucleotide variants (MNVs) differed greatly between replicates, probably caused by PCR-introduced bias. The total number of detected MNVs, calculated gene variability and mutational signature analysis, were unaffected by the sequencing run.
Conclusion: TaME-seq2 proved well suited for consensus sequence identification, and the detection of low-frequency viral genome variation and viral-chromosomal integrations. The repertoire of TaME-seq2 now encompasses seven HR-HPV types. Our goal is to further include all HR-HPV types in the TaME-seq2 repertoire. Moreover, with a minor modification of previously developed primers, the same method was successfully applied for the analysis of SARS-CoV-2 positive samples, implying the ease of adapting TaME-seq2 to other viruses.
Keywords: Genomics; HPV; Intra-host variation; Library preparation; NGS; SARS-CoV-2; Viral integration; Virology.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures


References
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
