

S-Adenosylmethionine: more than just a methyl donor
- PMID: 36891755
- PMCID: PMC10491745
- DOI: 10.1039/d2np00086e
S-Adenosylmethionine: more than just a methyl donor
Abstract
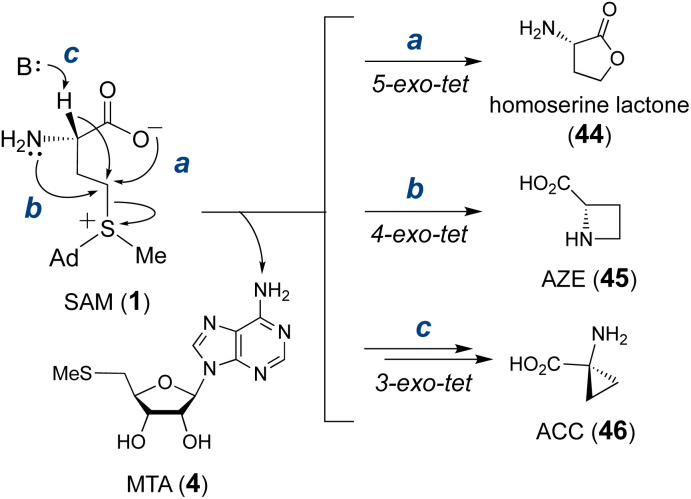
Covering: from 2000 up to the very early part of 2023S-Adenosyl-L-methionine (SAM) is a naturally occurring trialkyl sulfonium molecule that is typically associated with biological methyltransfer reactions. However, SAM is also known to donate methylene, aminocarboxypropyl, adenosyl and amino moieties during natural product biosynthetic reactions. The reaction scope is further expanded as SAM itself can be modified prior to the group transfer such that a SAM-derived carboxymethyl or aminopropyl moiety can also be transferred. Moreover, the sulfonium cation in SAM has itself been found to be critical for several other enzymatic transformations. Thus, while many SAM-dependent enzymes are characterized by a methyltransferase fold, not all of them are necessarily methyltransferases. Furthermore, other SAM-dependent enzymes do not possess such a structural feature suggesting diversification along different evolutionary lineages. Despite the biological versatility of SAM, it nevertheless parallels the chemistry of sulfonium compounds used in organic synthesis. The question thus becomes how enzymes catalyze distinct transformations via subtle differences in their active sites. This review summarizes recent advances in the discovery of novel SAM utilizing enzymes that rely on Lewis acid/base chemistry as opposed to radical mechanisms of catalysis. The examples are categorized based on the presence of a methyltransferase fold and the role played by SAM within the context of known sulfonium chemistry.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures










































References
-
- Blum M. Chang H.-Y. Chuguransky S. Grego T. Kandasaamy S. Mitchell A. Nuka G. Paysan-Lafosse T. Qureshi M. Raj S. Richardson L. Salazar G. A. Williams L. Bork P. Bridge A. Gough J. Haft D. H. Letunic I. Marchler-Bauer A. Mi H. Natale D. A. Necci M. Orengo C. A. Pandurangan A. P. Rivoire C. Sigrist C. J. A. Sillitoe I. Thanki N. Thomas P. D. Tosatto S. C. E. Wu C. H. Bateman A. Finn R. D. Nucleic Acids Res. 2021;49:D344–D354. doi: 10.1093/nar/gkaa977. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources