

KMT2D acetylation by CREBBP reveals a cooperative functional interaction at enhancers in normal and malignant germinal center B cells
- PMID: 36893259
- PMCID: PMC10089214
- DOI: 10.1073/pnas.2218330120
KMT2D acetylation by CREBBP reveals a cooperative functional interaction at enhancers in normal and malignant germinal center B cells
Abstract
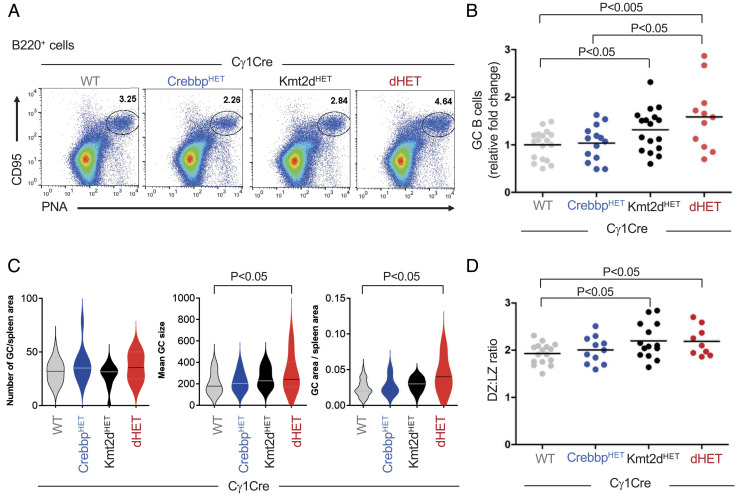
Heterozygous inactivating mutations of the KMT2D methyltransferase and the CREBBP acetyltransferase are among the most common genetic alterations in B cell lymphoma and co-occur in 40 to 60% of follicular lymphoma (FL) and 30% of EZB/C3 diffuse large B cell lymphoma (DLBCL) cases, suggesting they may be coselected. Here, we show that combined germinal center (GC)-specific haploinsufficiency of Crebbp and Kmt2d synergizes in vivo to promote the expansion of abnormally polarized GCs, a common preneoplastic event. These enzymes form a biochemical complex on select enhancers/superenhancers that are critical for the delivery of immune signals in the GC light zone and are only corrupted upon dual Crebbp/Kmt2d loss, both in mouse GC B cells and in human DLBCL. Moreover, CREBBP directly acetylates KMT2D in GC-derived B cells, and, consistently, its inactivation by FL/DLBCL-associated mutations abrogates its ability to catalyze KMT2D acetylation. Genetic and pharmacologic loss of CREBBP and the consequent decrease in KMT2D acetylation lead to reduced levels of H3K4me1, supporting a role for this posttranslational modification in modulating KMT2D activity. Our data identify a direct biochemical and functional interaction between CREBBP and KMT2D in the GC, with implications for their role as tumor suppressors in FL/DLBCL and for the development of precision medicine approaches targeting enhancer defects induced by their combined loss.
Keywords: B cell lymphoma; CREBBP; KMT2D; acetylation; germinal center.
Conflict of interest statement
The authors have organizational affiliations to disclose, ASTRA ZENECA: Consulting; NEOGENOMICS: Advisory Board Member, the authors have research support to disclose, ASTRA ZENECA: Research Award CU20-0758.
Figures







References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
