

Cytotoxic CD161-CD8+ TEMRA cells contribute to the pathogenesis of systemic lupus erythematosus
- PMID: 36893588
- PMCID: PMC10011749
- DOI: 10.1016/j.ebiom.2023.104507
Cytotoxic CD161-CD8+ TEMRA cells contribute to the pathogenesis of systemic lupus erythematosus
Abstract
Background: Systemic lupus erythematosus (SLE) is a prototypical autoimmune disease affecting multiple organs and tissues with high cellular heterogeneity. CD8+ T cell activity is involved in the SLE pathogenesis. However, the cellular heterogeneity and the underlying mechanisms of CD8+ T cells in SLE remain to be identified.
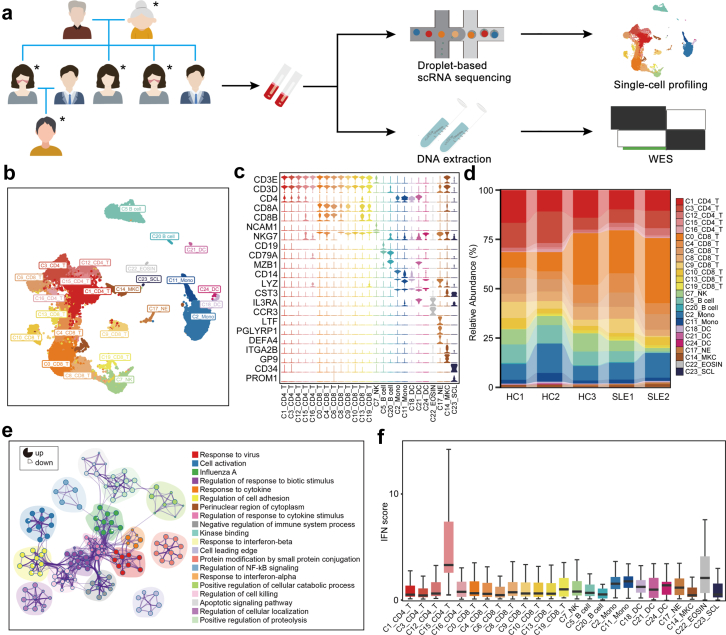
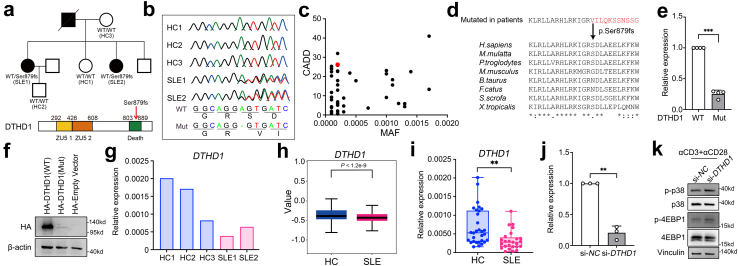
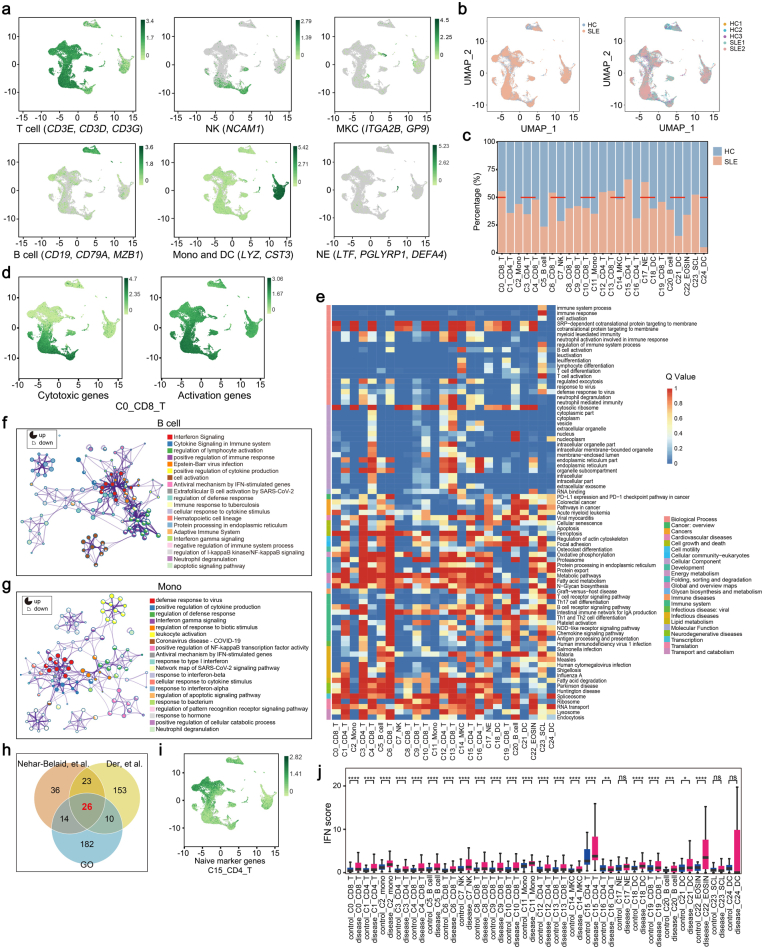
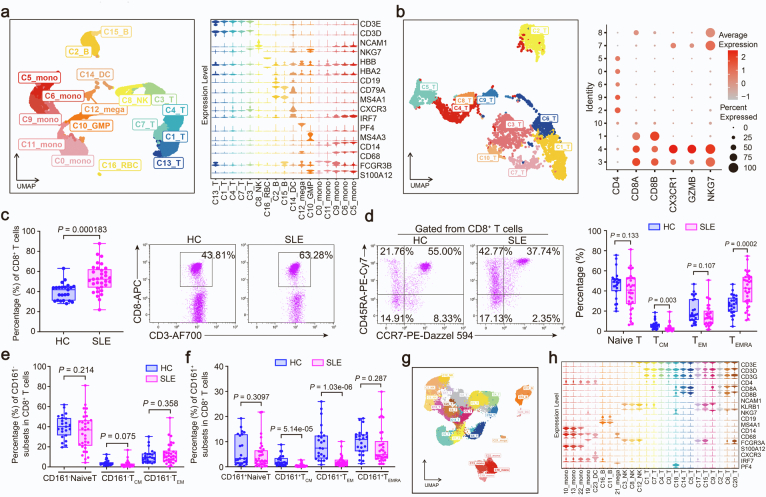
Methods: Single-cell RNA sequencing (scRNA-seq) of PBMCs from a SLE family pedigree (including 3 HCs and 2 SLE patients) was performed to identify the SLE-associated CD8+ T cell subsets. Flow cytometry analysis of a SLE cohort (including 23 HCs and 33 SLE patients), qPCR analysis of another SLE cohort (including 30 HCs and 25 SLE patients) and public scRNA-seq datasets of autoimmune diseases were employed to validate the finding. Whole-exome sequencing (WES) of this SLE family pedigree was used to investigate the genetic basis in dysregulation of CD8+ T cell subsets identified in this study. Co-culture experiments were performed to analyze the activity of CD8+ T cells.
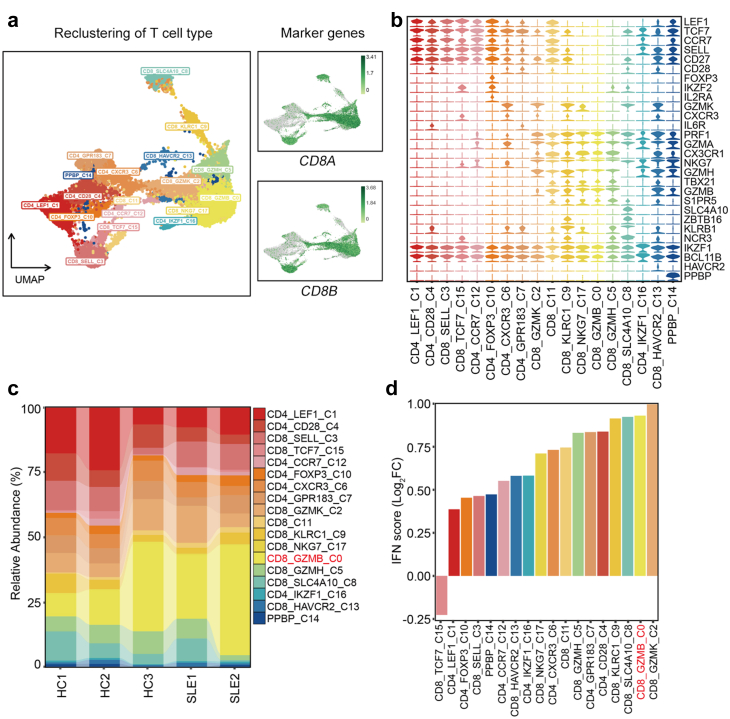
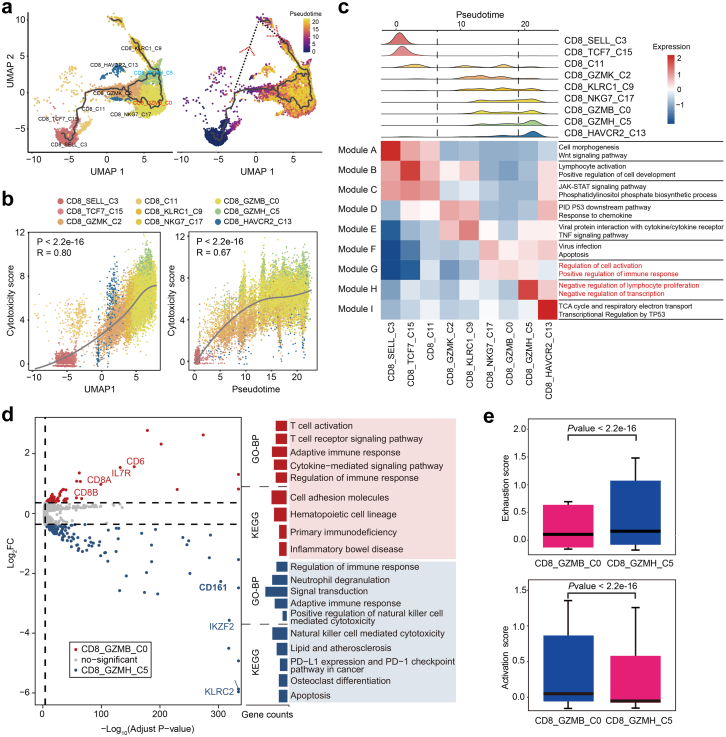
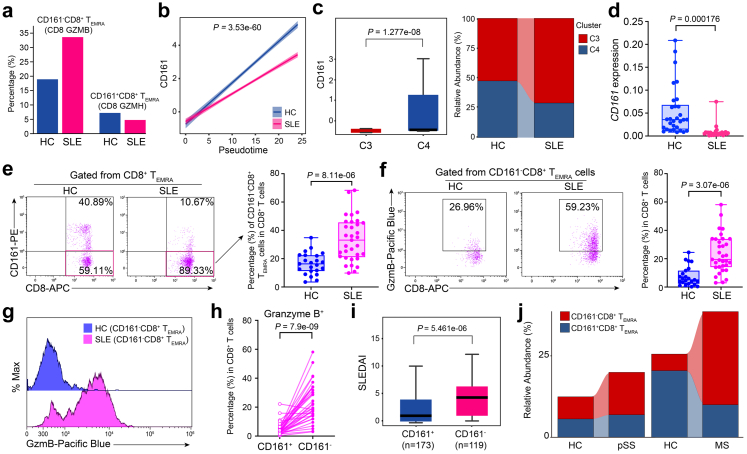
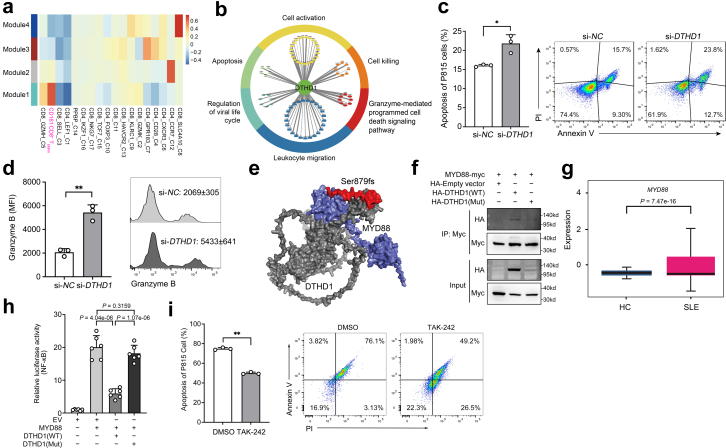
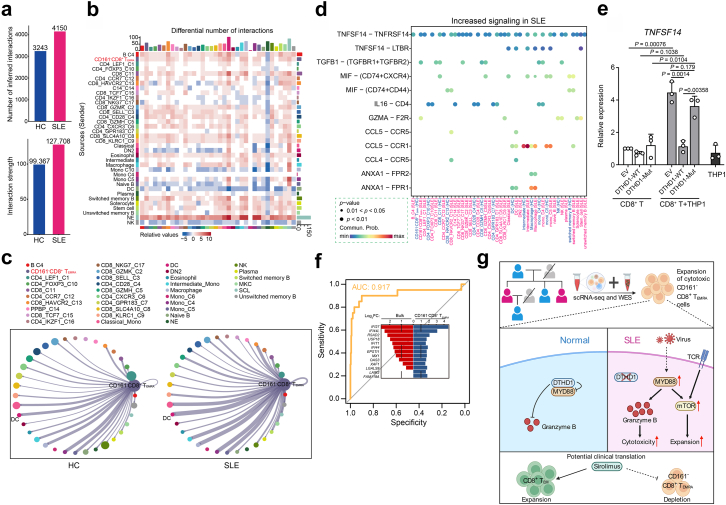
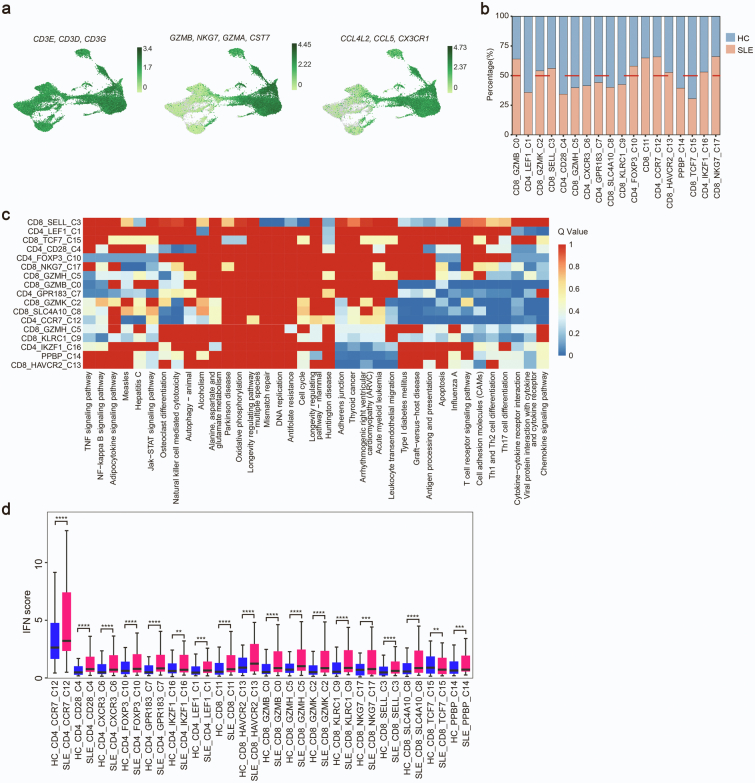
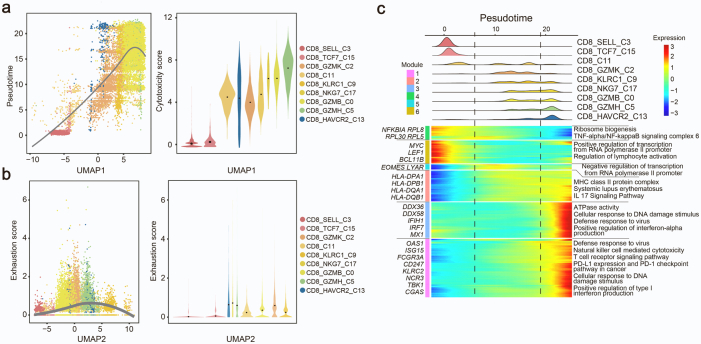
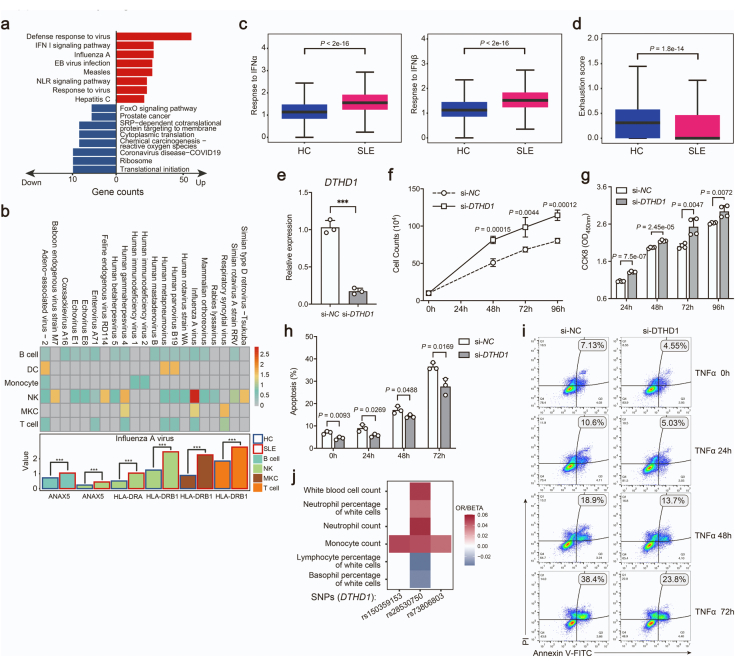
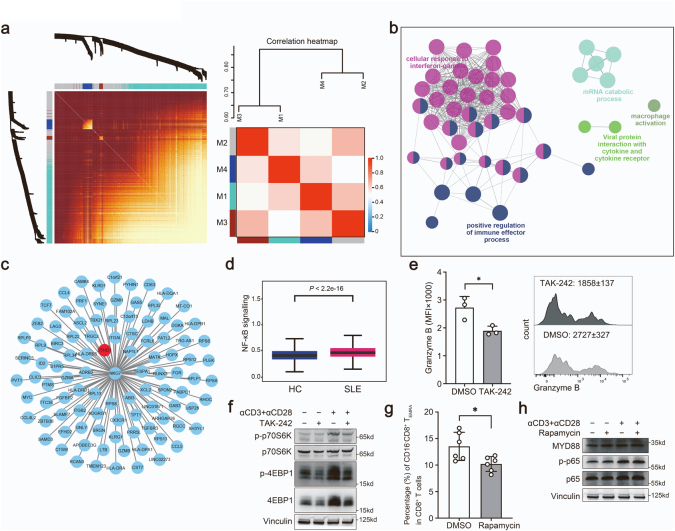
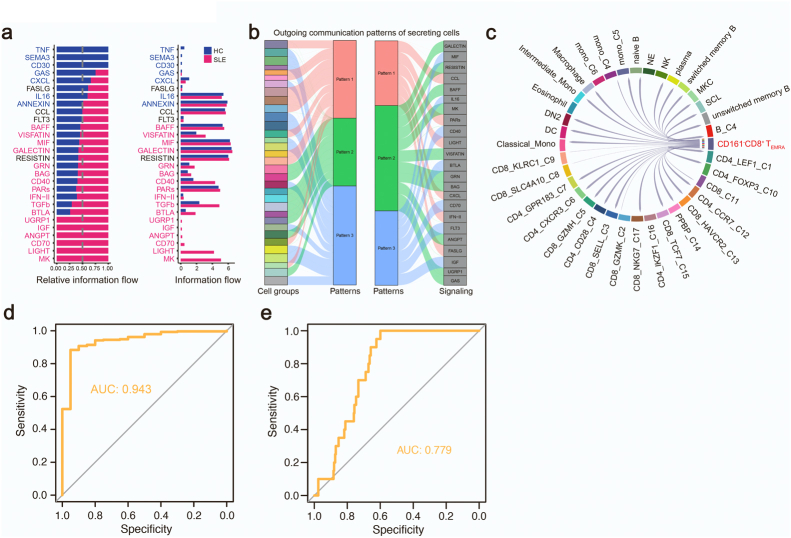
Findings: We elucidated the cellular heterogeneity of SLE and identified a new highly cytotoxic CD8+ T cell subset, CD161-CD8+ TEMRA cell subpopulation, which was remarkably increased in SLE patients. Meanwhile, we discovered a close correlation between mutation of DTHD1 and the abnormal accumulation of CD161-CD8+ TEMRA cells in SLE. DTHD1 interacted with MYD88 to suppress its activity in T cells and DTHD1 mutation promoted MYD88-dependent pathway and subsequently increased the proliferation and cytotoxicity of CD161-CD8+ TEMRA cells. Furthermore, the differentially expressed genes in CD161-CD8+ TEMRA cells displayed a strong out-of-sample prediction for case-control status of SLE.
Interpretation: This study identified DTHD1-associated expansion of CD161-CD8+ TEMRA cell subpopulation is critical for SLE. Our study highlights genetic association and cellular heterogeneity of SLE pathogenesis and provides a mechanistical insight into the diagnosis and treatment of SLE.
Fundings: Stated in the Acknowledgements section of the manuscript.
Keywords: CD8(+) T cell subset; DTHD1; Genetic variant; MYD88; Systemic lupus erythematosus; Whole-exome sequencing; scRNA-seq.
Copyright © 2023 The Author(s). Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare that they have no competing interests.
Figures
References
-
- Tsokos G.C. Systemic lupus erythematosus. N Engl J Med. 2011;365(22):2110–2121. - PubMed
-
- Dorner T., Furie R. Novel paradigms in systemic lupus erythematosus. Lancet. 2019;393(10188):2344–2358. - PubMed
-
- Kaul A., Gordon C., Crow M.K., et al. Systemic lupus erythematosus. Nat Rev Dis Primers. 2016;2 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
