

Mechanisms of pathogenicity in the hypertrophic cardiomyopathy-associated TPM1 variant S215L
- PMID: 36896133
- PMCID: PMC9991458
- DOI: 10.1093/pnasnexus/pgad011
Mechanisms of pathogenicity in the hypertrophic cardiomyopathy-associated TPM1 variant S215L
Abstract
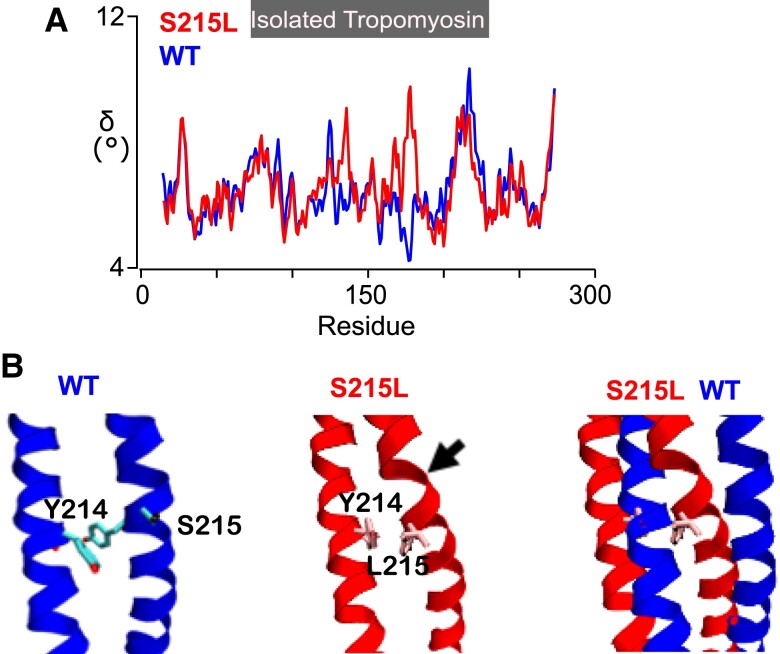
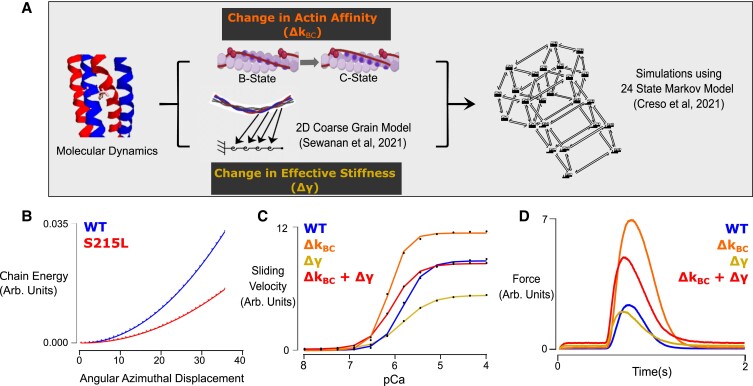
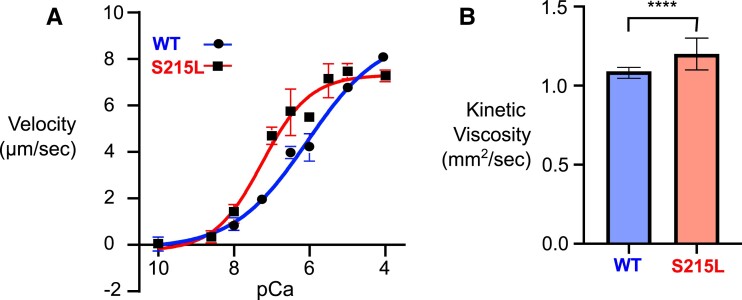
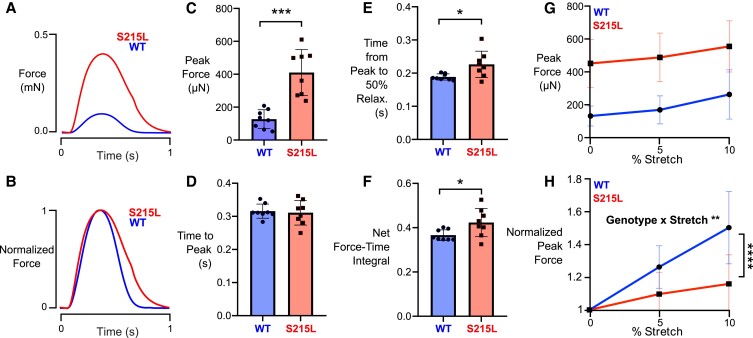
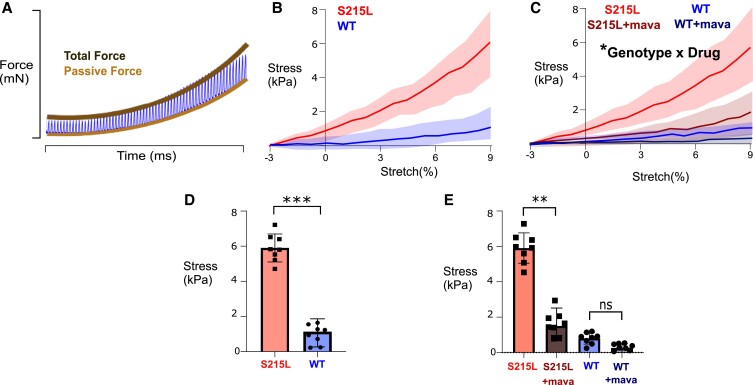
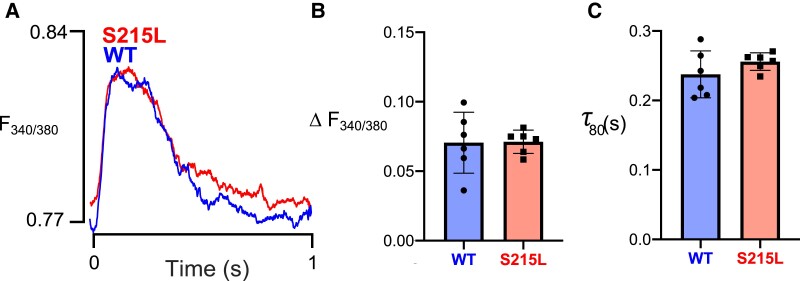
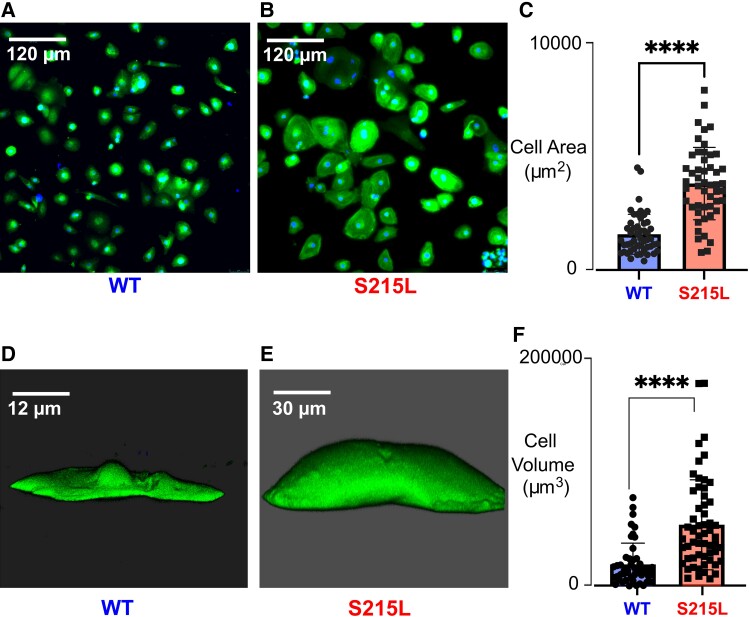
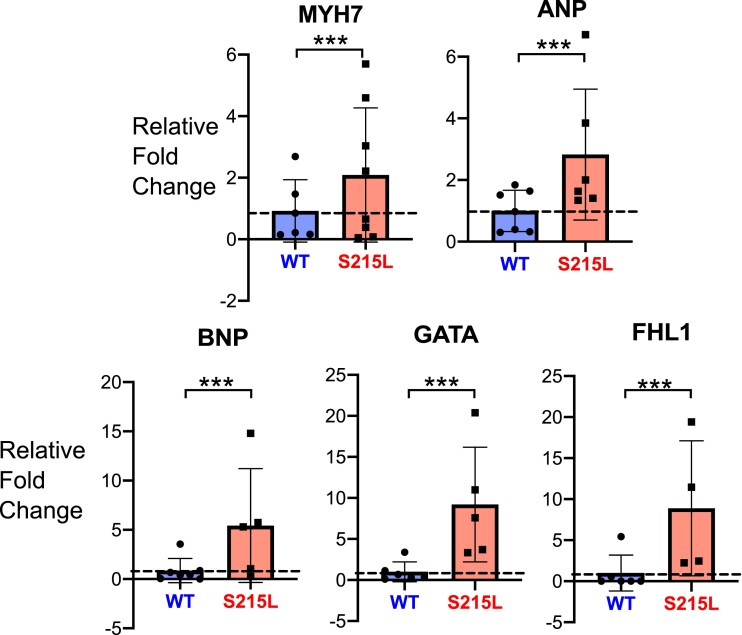
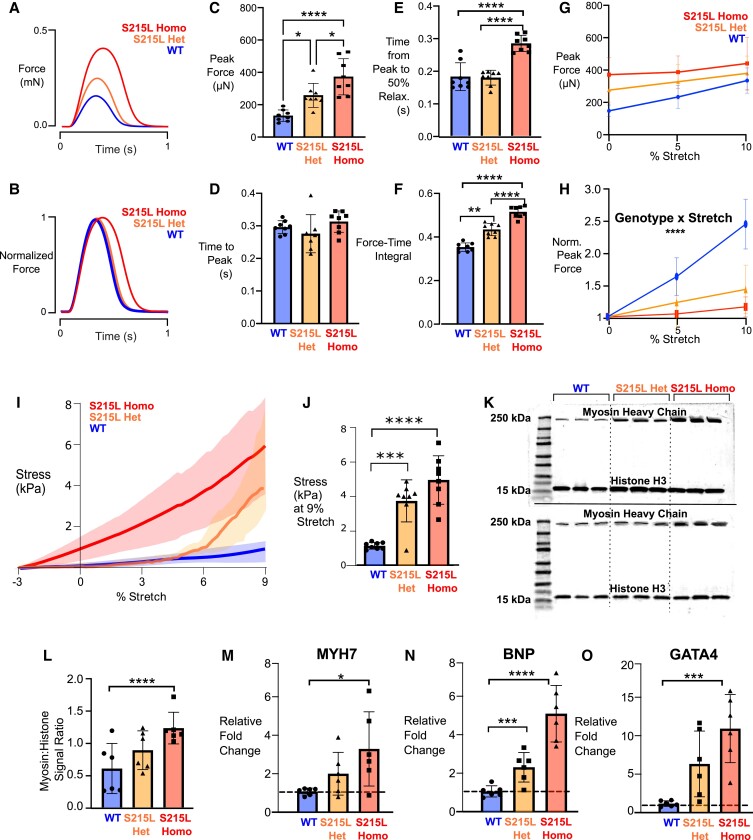
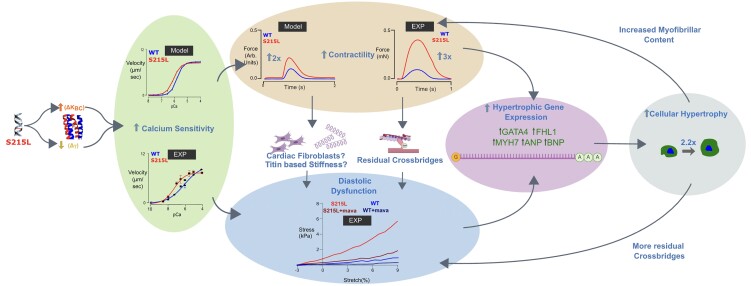
Hypertrophic cardiomyopathy (HCM) is an inherited disorder often caused by mutations to sarcomeric genes. Many different HCM-associated TPM1 mutations have been identified but they vary in their degrees of severity, prevalence, and rate of disease progression. The pathogenicity of many TPM1 variants detected in the clinical population remains unknown. Our objective was to employ a computational modeling pipeline to assess pathogenicity of one such variant of unknown significance, TPM1 S215L, and validate predictions using experimental methods. Molecular dynamic simulations of tropomyosin on actin suggest that the S215L significantly destabilizes the blocked regulatory state while increasing flexibility of the tropomyosin chain. These changes were quantitatively represented in a Markov model of thin-filament activation to infer the impacts of S215L on myofilament function. Simulations of in vitro motility and isometric twitch force predicted that the mutation would increase Ca2+ sensitivity and twitch force while slowing twitch relaxation. In vitro motility experiments with thin filaments containing TPM1 S215L revealed higher Ca2+ sensitivity compared with wild type. Three-dimensional genetically engineered heart tissues expressing TPM1 S215L exhibited hypercontractility, upregulation of hypertrophic gene markers, and diastolic dysfunction. These data form a mechanistic description of TPM1 S215L pathogenicity that starts with disruption of the mechanical and regulatory properties of tropomyosin, leading thereafter to hypercontractility and finally induction of a hypertrophic phenotype. These simulations and experiments support the classification of S215L as a pathogenic mutation and support the hypothesis that an inability to adequately inhibit actomyosin interactions is the mechanism whereby thin-filament mutations cause HCM.
Keywords: engineered heart tissue; hypertrophic cardiomyopathy; tropomyosin.
© The Author(s) 2023. Published by Oxford University Press on behalf of National Academy of Sciences.
Figures
References
-
- Yotti R, Seidman CE, Seidman JG. 2019. Advances in the genetic basis and pathogenesis of sarcomere cardiomyopathies. Annu Rev Genomics Hum Genet. 20:129–153. - PubMed
-
- Kaltman JR, et al. . 2011. Screening for sudden cardiac death in the young: report from a national heart, lung, and blood institute working group. Circulation 123(17):1911–1918. - PubMed
-
- Risgaard B. 2016. Sudden cardiac death: a nationwide cohort study among the young. Dan Med J. 63(12):B5321. - PubMed
-
- Lynge TH, et al. . 2016. Cardiac symptoms before sudden cardiac death caused by hypertrophic cardiomyopathy: a nationwide study among the young in Denmark. Europace 18(12):1801–1808. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
