

Blocking Store-Operated Ca2+ Entry to Protect HL-1 Cardiomyocytes from Epirubicin-Induced Cardiotoxicity
- PMID: 36899859
- PMCID: PMC10000558
- DOI: 10.3390/cells12050723
Blocking Store-Operated Ca2+ Entry to Protect HL-1 Cardiomyocytes from Epirubicin-Induced Cardiotoxicity
Abstract
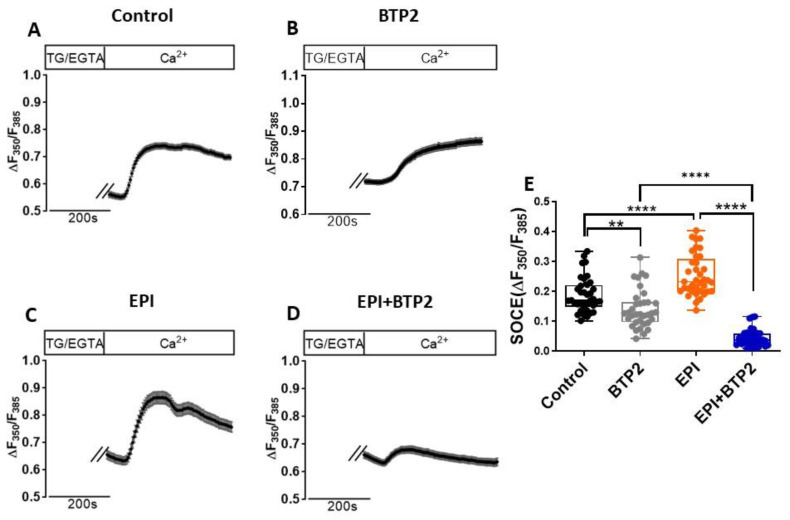
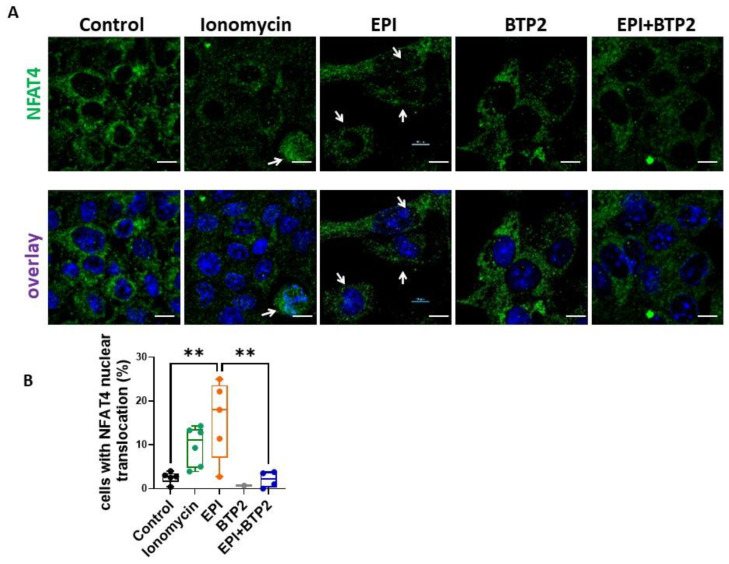
Epirubicin (EPI) is one of the most widely used anthracycline chemotherapy drugs, yet its cardiotoxicity severely limits its clinical application. Altered intracellular Ca2+ homeostasis has been shown to contribute to EPI-induced cell death and hypertrophy in the heart. While store-operated Ca2+ entry (SOCE) has recently been linked with cardiac hypertrophy and heart failure, its role in EPI-induced cardiotoxicity remains unknown. Using a publicly available RNA-seq dataset of human iPSC-derived cardiomyocytes, gene analysis showed that cells treated with 2 µM EPI for 48 h had significantly reduced expression of SOCE machinery genes, e.g., Orai1, Orai3, TRPC3, TRPC4, Stim1, and Stim2. Using HL-1, a cardiomyocyte cell line derived from adult mouse atria, and Fura-2, a ratiometric Ca2+ fluorescent dye, this study confirmed that SOCE was indeed significantly reduced in HL-1 cells treated with EPI for 6 h or longer. However, HL-1 cells presented increased SOCE as well as increased reactive oxygen species (ROS) production at 30 min after EPI treatment. EPI-induced apoptosis was evidenced by disruption of F-actin and increased cleavage of caspase-3 protein. The HL-1 cells that survived to 24 h after EPI treatment demonstrated enlarged cell sizes, up-regulated expression of brain natriuretic peptide (a hypertrophy marker), and increased NFAT4 nuclear translocation. Treatment by BTP2, a known SOCE blocker, decreased the initial EPI-enhanced SOCE, rescued HL-1 cells from EPI-induced apoptosis, and reduced NFAT4 nuclear translocation and hypertrophy. This study suggests that EPI may affect SOCE in two phases: the initial enhancement phase and the following cell compensatory reduction phase. Administration of a SOCE blocker at the initial enhancement phase may protect cardiomyocytes from EPI-induced toxicity and hypertrophy.
Keywords: NFAT4; anthracycline; apoptosis; cardiac hypertrophy; chemotherapy; reactive oxygen species (ROS); store-operated Ca2+ entry (SOCE).
Conflict of interest statement
The authors declare no conflict of interest.
Figures





References
-
- World Health Organization WHO Model List of Essential Medicines—22nd List. 2021. [(accessed on 30 September 2021)]. Available online: https://www.who.int/publications/i/item/WHO-MHP-HPS-EML-2021.02.
-
- Zamorano J.L., Lancellotti P., Rodriguez Munoz D., Aboyans V., Asteggiano R., Galderisi M., Habib G., Lenihan D.J., Lip G.Y.H., Lyon A.R., et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC) Eur. Heart J. 2016;37:2768–2801. doi: 10.1093/eurheartj/ehw211. - DOI - PubMed
-
- Cardinale D., Colombo A., Lamantia G., Colombo N., Civelli M., De Giacomi G., Rubino M., Veglia F., Fiorentini C., Cipolla C.M. Anthracycline-induced cardiomyopathy: Clinical relevance and response to pharmacologic therapy. J. Am. Coll. Cardiol. 2010;55:213–220. doi: 10.1016/j.jacc.2009.03.095. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
