

The translational landscape of bread wheat during grain development
- PMID: 36905284
- PMCID: PMC10226598
- DOI: 10.1093/plcell/koad075
The translational landscape of bread wheat during grain development
Abstract
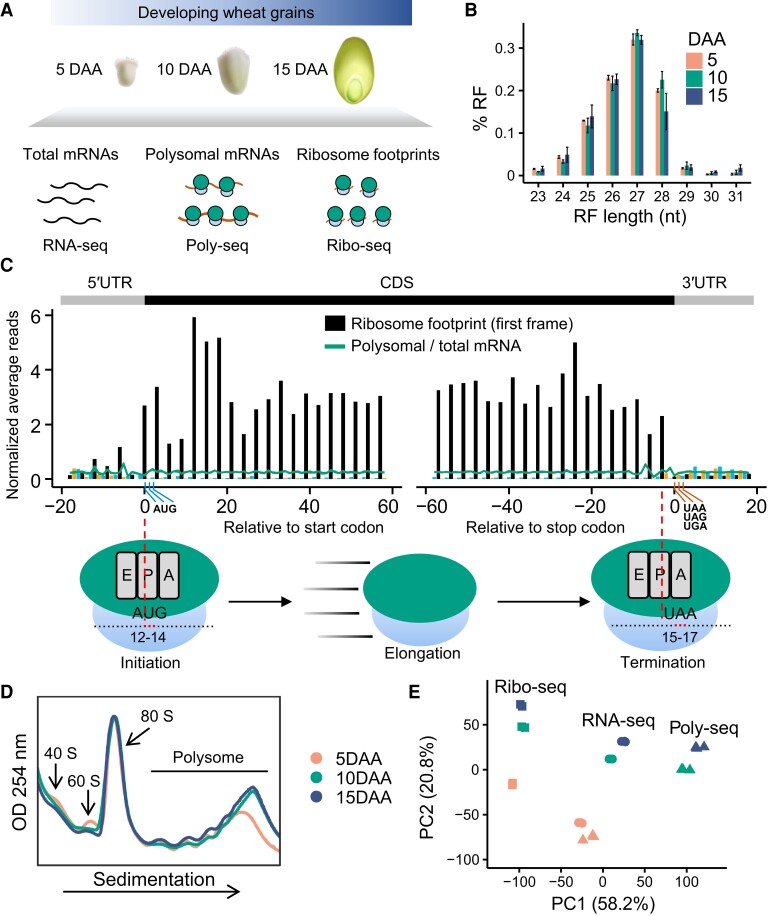
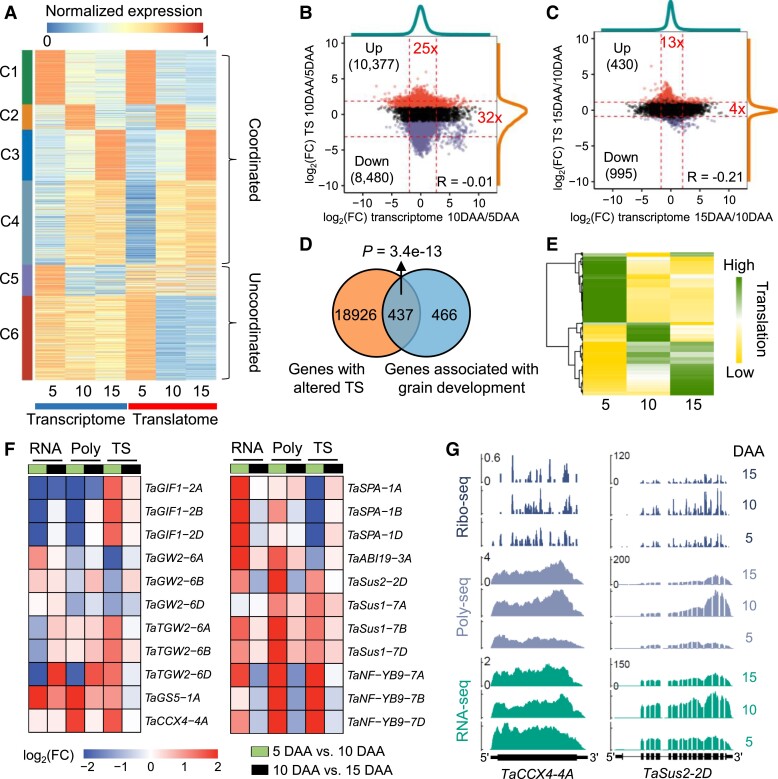
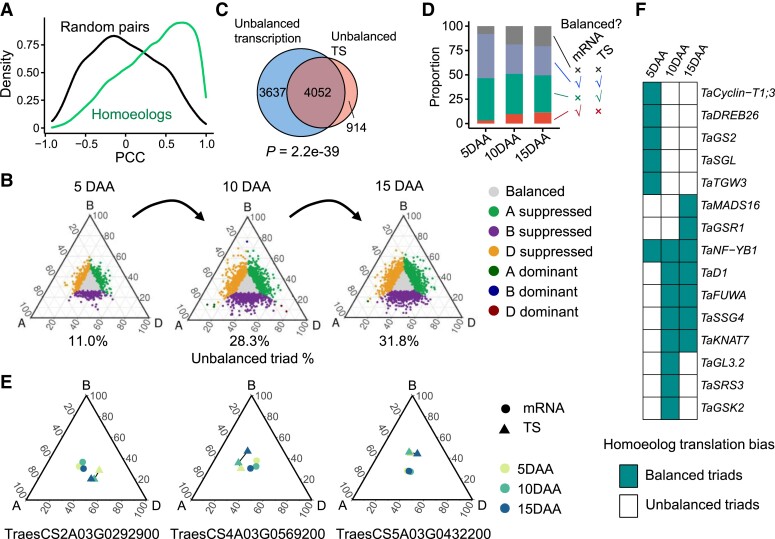
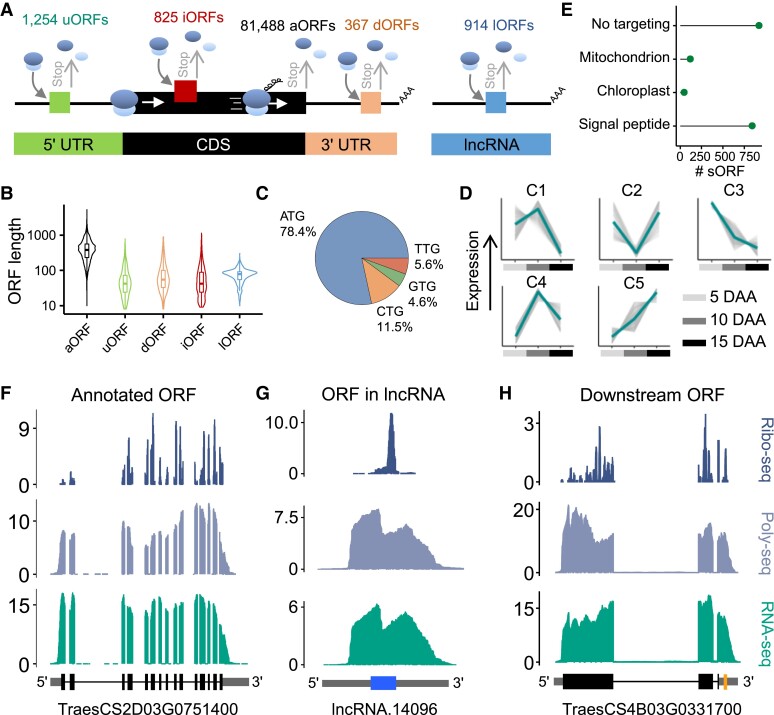
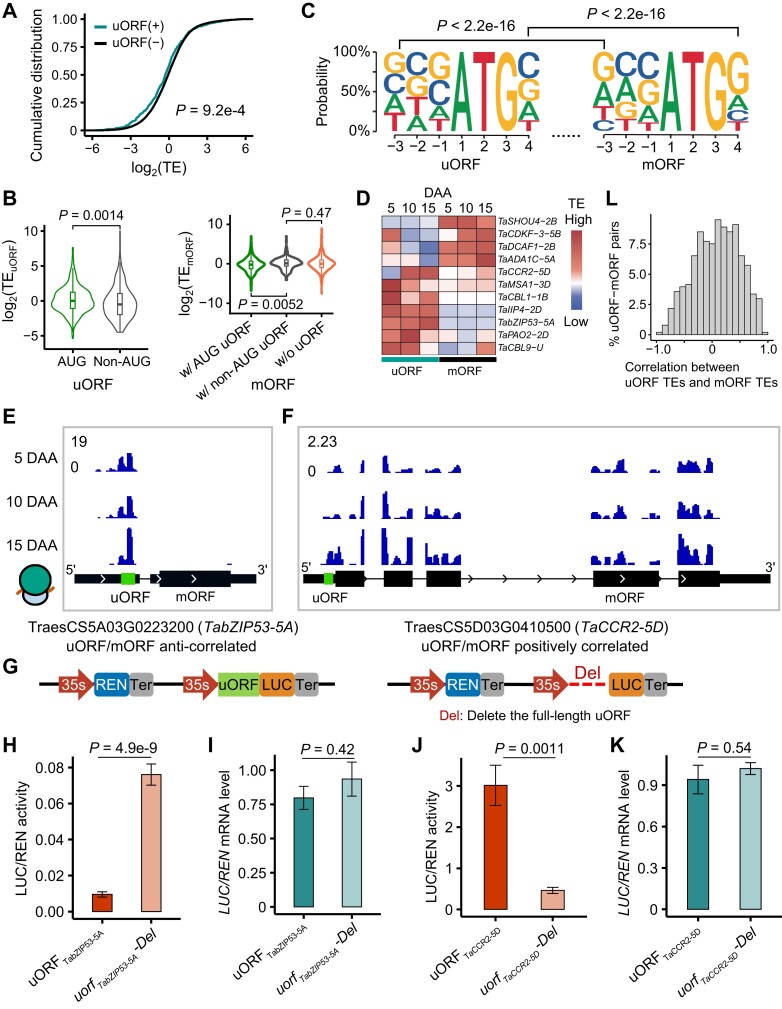
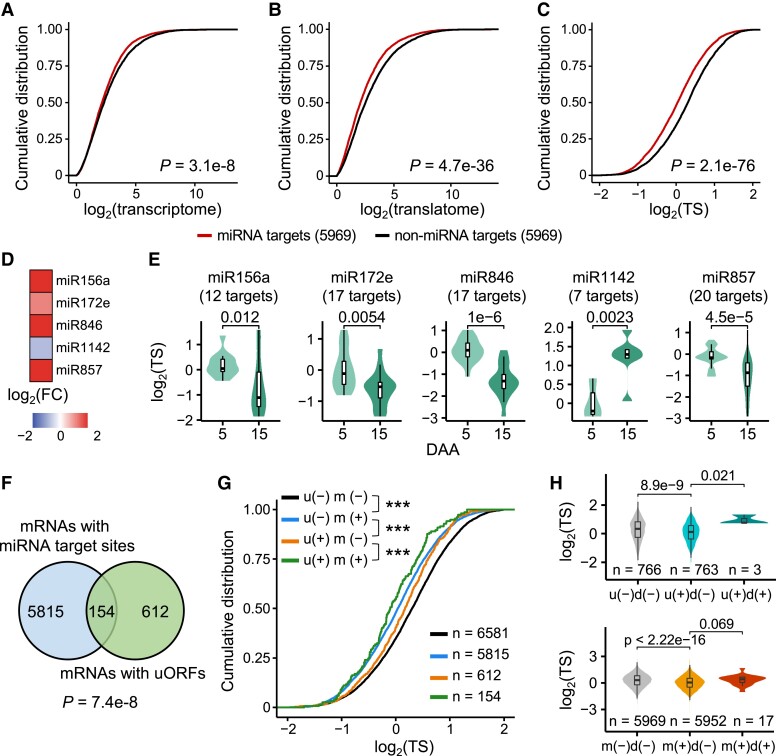
The dynamics of gene expression in crop grains has typically been investigated at the transcriptional level. However, this approach neglects translational regulation, a widespread mechanism that rapidly modulates gene expression to increase the plasticity of organisms. Here, we performed ribosome profiling and polysome profiling to obtain a comprehensive translatome data set of developing bread wheat (Triticum aestivum) grains. We further investigated the genome-wide translational dynamics during grain development, revealing that the translation of many functional genes is modulated in a stage-specific manner. The unbalanced translation between subgenomes is pervasive, which increases the expression flexibility of allohexaploid wheat. In addition, we uncovered widespread previously unannotated translation events, including upstream open reading frames (uORFs), downstream open reading frames (dORFs), and open reading frames (ORFs) in long noncoding RNAs, and characterized the temporal expression dynamics of small ORFs. We demonstrated that uORFs act as cis-regulatory elements that can repress or even enhance the translation of mRNAs. Gene translation may be combinatorially modulated by uORFs, dORFs, and microRNAs. In summary, our study presents a translatomic resource that provides a comprehensive and detailed overview of the translational regulation in developing bread wheat grains. This resource will facilitate future crop improvements for optimal yield and quality.
© American Society of Plant Biologists 2023. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Conflict of interest statement
Conflict of interest statement. None declared.
Figures
Comment in
-
Whole grain: A genome-wide landscape of translational regulation during bread wheat development.Plant Cell. 2023 May 29;35(6):1621-1622. doi: 10.1093/plcell/koad078. Plant Cell. 2023. PMID: 36916512 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
