

Tumor cell plasticity in targeted therapy-induced resistance: mechanisms and new strategies
- PMID: 36906600
- PMCID: PMC10008648
- DOI: 10.1038/s41392-023-01383-x
Tumor cell plasticity in targeted therapy-induced resistance: mechanisms and new strategies
Abstract
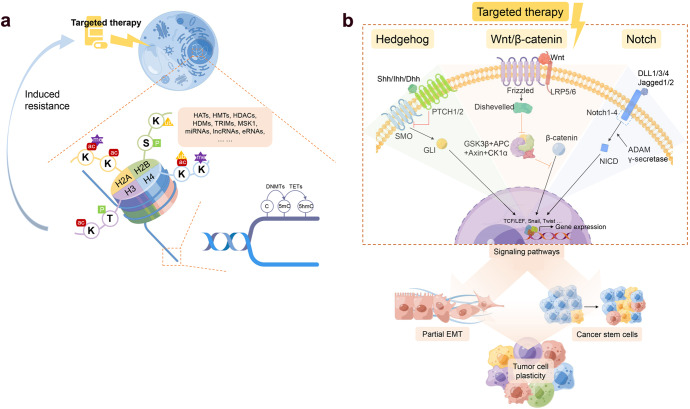
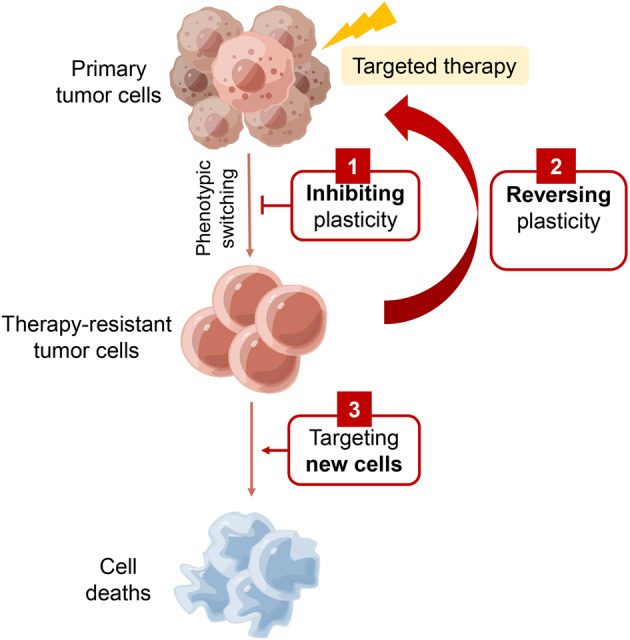
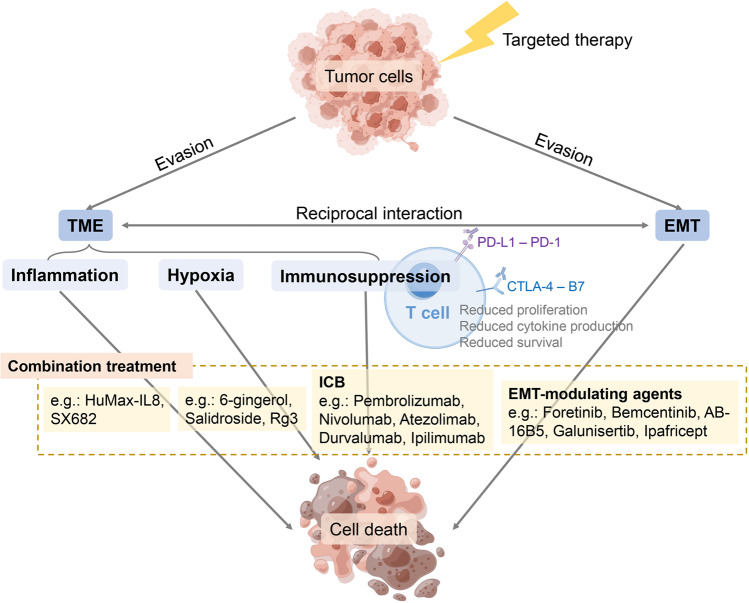
Despite the success of targeted therapies in cancer treatment, therapy-induced resistance remains a major obstacle to a complete cure. Tumor cells evade treatments and relapse via phenotypic switching driven by intrinsic or induced cell plasticity. Several reversible mechanisms have been proposed to circumvent tumor cell plasticity, including epigenetic modifications, regulation of transcription factors, activation or suppression of key signaling pathways, as well as modification of the tumor environment. Epithelial-to-mesenchymal transition, tumor cell and cancer stem cell formation also serve as roads towards tumor cell plasticity. Corresponding treatment strategies have recently been developed that either target plasticity-related mechanisms or employ combination treatments. In this review, we delineate the formation of tumor cell plasticity and its manipulation of tumor evasion from targeted therapy. We discuss the non-genetic mechanisms of targeted drug-induced tumor cell plasticity in various types of tumors and provide insights into the contribution of tumor cell plasticity to acquired drug resistance. New therapeutic strategies such as inhibition or reversal of tumor cell plasticity are also presented. We also discuss the multitude of clinical trials that are ongoing worldwide with the intention of improving clinical outcomes. These advances provide a direction for developing novel therapeutic strategies and combination therapy regimens that target tumor cell plasticity.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


References
-
- Waddington CH. Embryology, epigenetics and biogenetics. Nature. 1956;177:1241–1241. doi: 10.1038/1771241a0. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
