

Interpretable and context-free deconvolution of multi-scale whole transcriptomic data with UniCell deconvolve
- PMID: 36906603
- PMCID: PMC10008582
- DOI: 10.1038/s41467-023-36961-8
Interpretable and context-free deconvolution of multi-scale whole transcriptomic data with UniCell deconvolve
Abstract
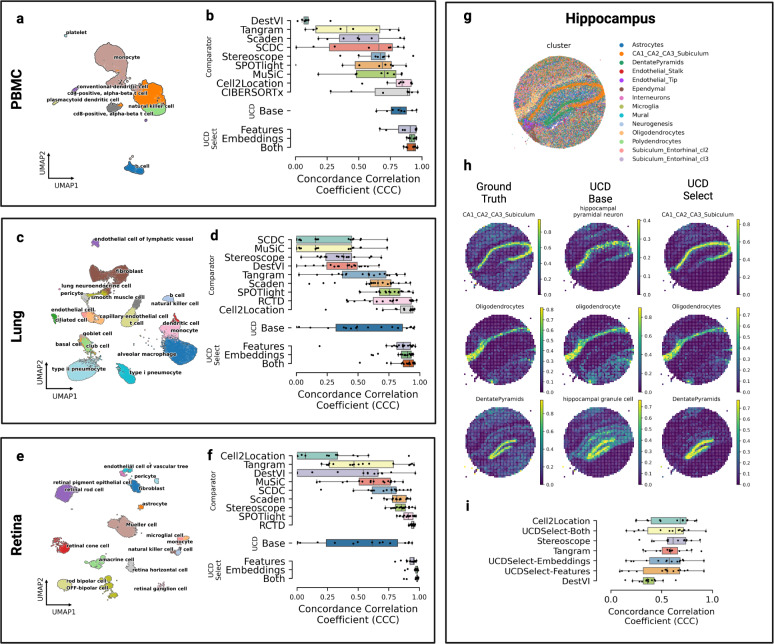
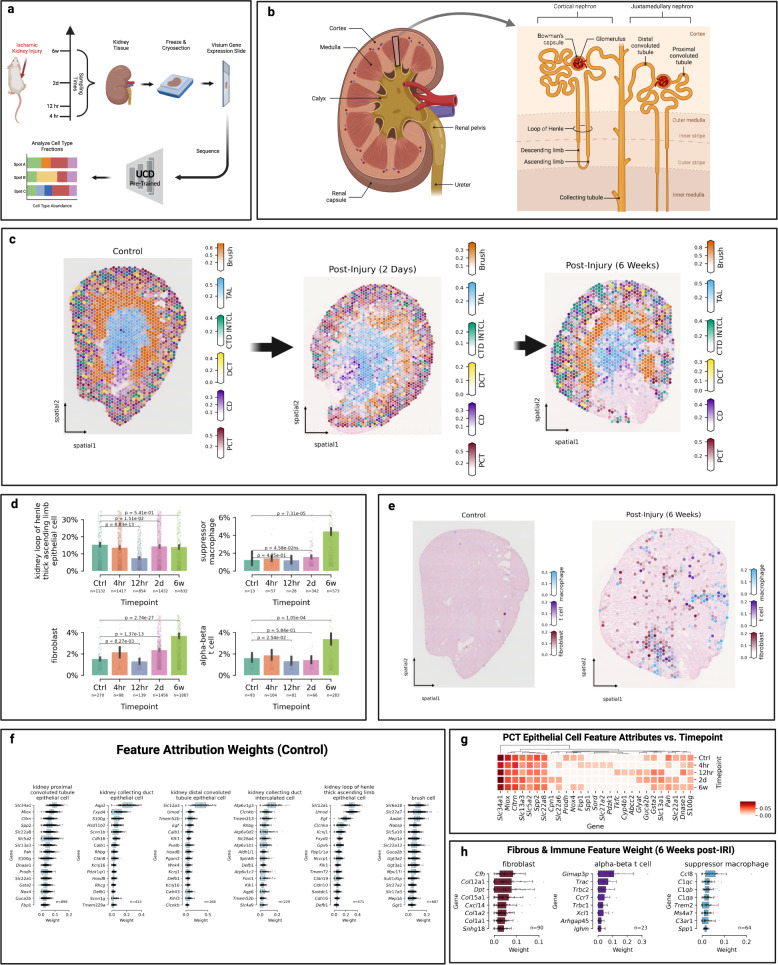
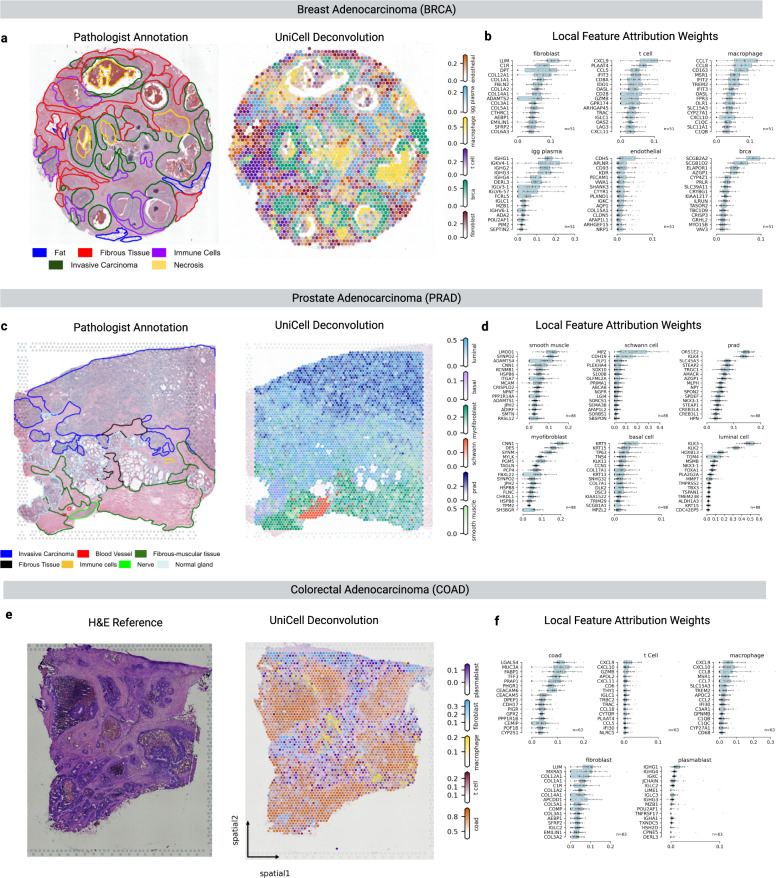
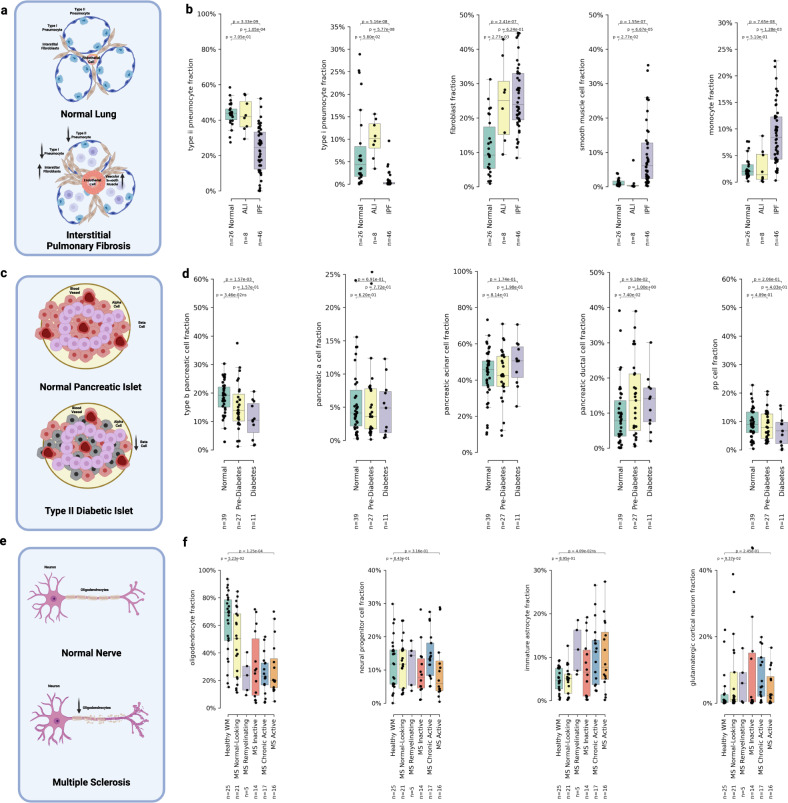
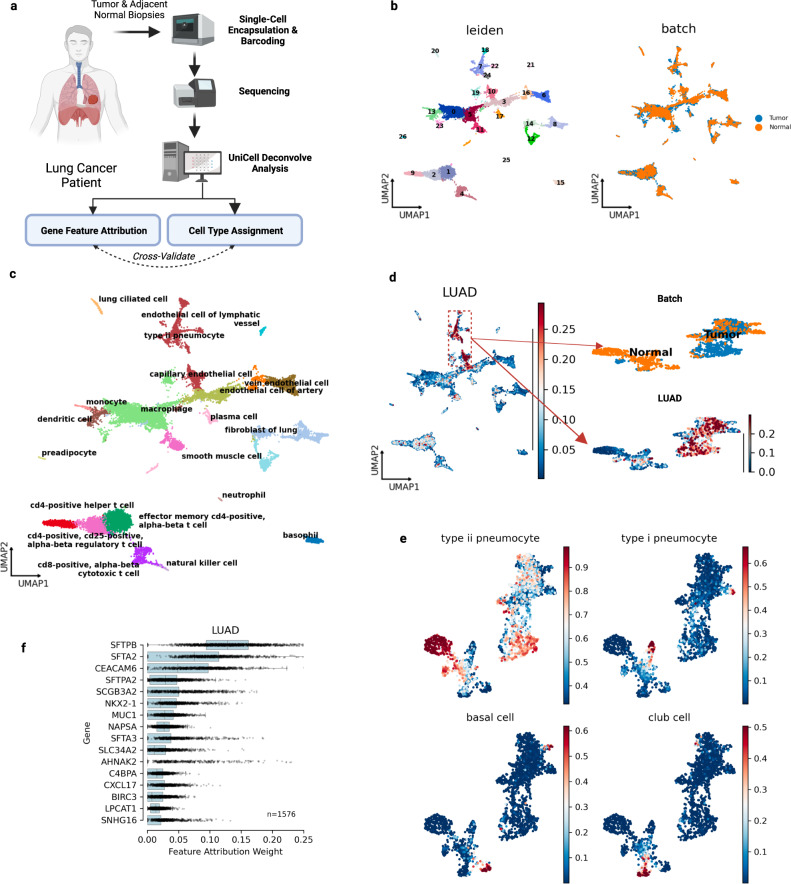
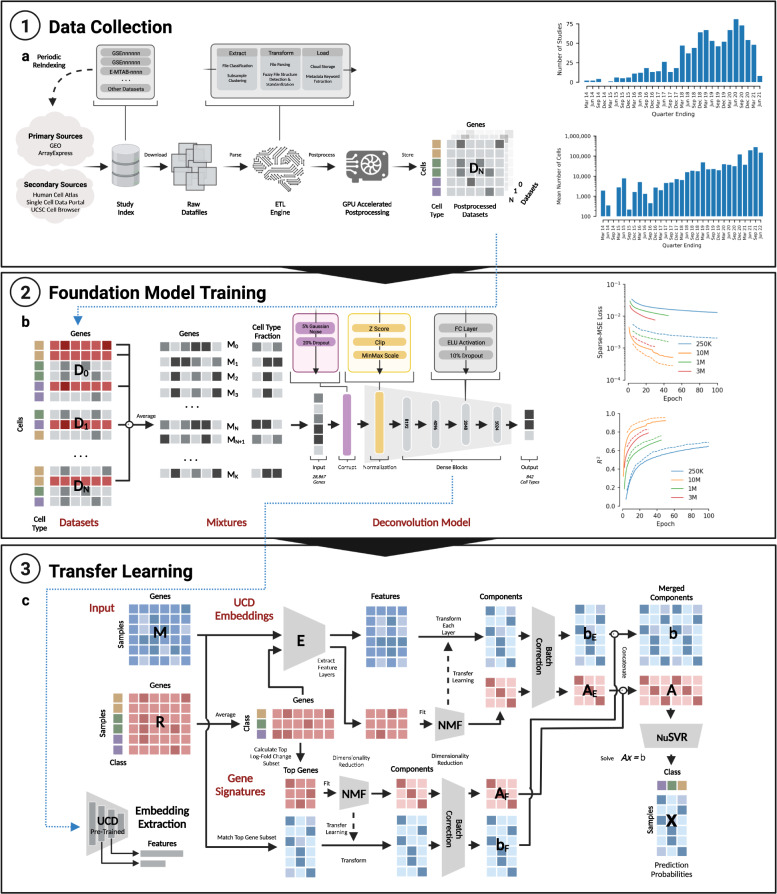
We introduce UniCell: Deconvolve Base (UCDBase), a pre-trained, interpretable, deep learning model to deconvolve cell type fractions and predict cell identity across Spatial, bulk-RNA-Seq, and scRNA-Seq datasets without contextualized reference data. UCD is trained on 10 million pseudo-mixtures from a fully-integrated scRNA-Seq training database comprising over 28 million annotated single cells spanning 840 unique cell types from 898 studies. We show that our UCDBase and transfer-learning models achieve comparable or superior performance on in-silico mixture deconvolution to existing, reference-based, state-of-the-art methods. Feature attribute analysis uncovers gene signatures associated with cell-type specific inflammatory-fibrotic responses in ischemic kidney injury, discerns cancer subtypes, and accurately deconvolves tumor microenvironments. UCD identifies pathologic changes in cell fractions among bulk-RNA-Seq data for several disease states. Applied to lung cancer scRNA-Seq data, UCD annotates and distinguishes normal from cancerous cells. Overall, UCD enhances transcriptomic data analysis, aiding in assessment of cellular and spatial context.
© 2023. The Author(s).
Conflict of interest statement
D.C., R.B., and R.S. declare no competing interests. RS is a consultant/advisor as of July 2022 at GeneDx, a company with no direct relationships to the present work.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
