

The first genetic landscape of inherited retinal dystrophies in Portuguese patients identifies recurrent homozygous mutations as a frequent cause of pathogenesis
- PMID: 36909829
- PMCID: PMC10003751
- DOI: 10.1093/pnasnexus/pgad043
The first genetic landscape of inherited retinal dystrophies in Portuguese patients identifies recurrent homozygous mutations as a frequent cause of pathogenesis
Abstract
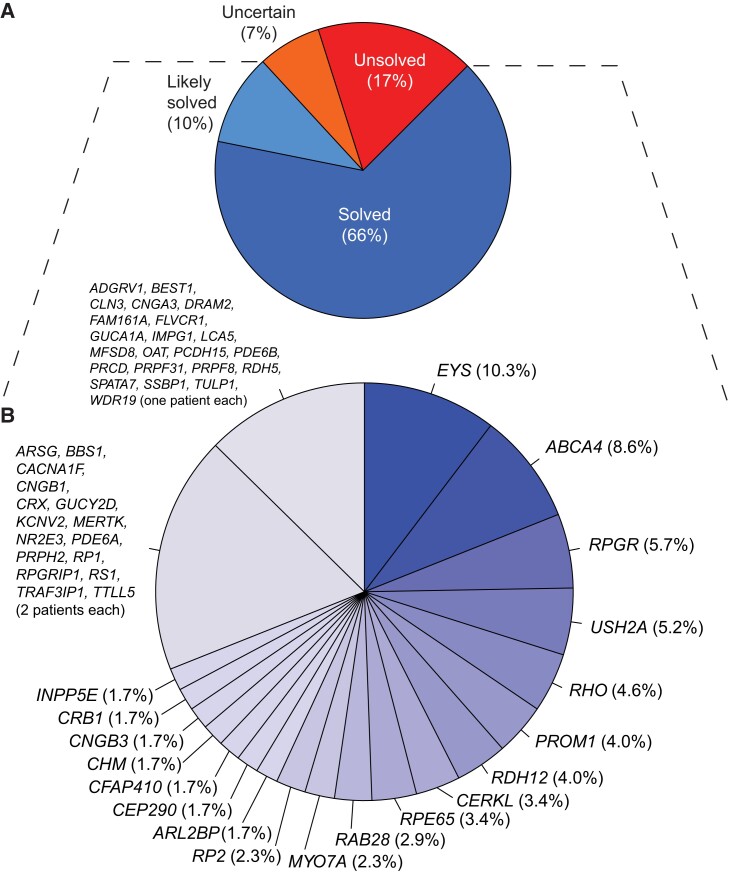
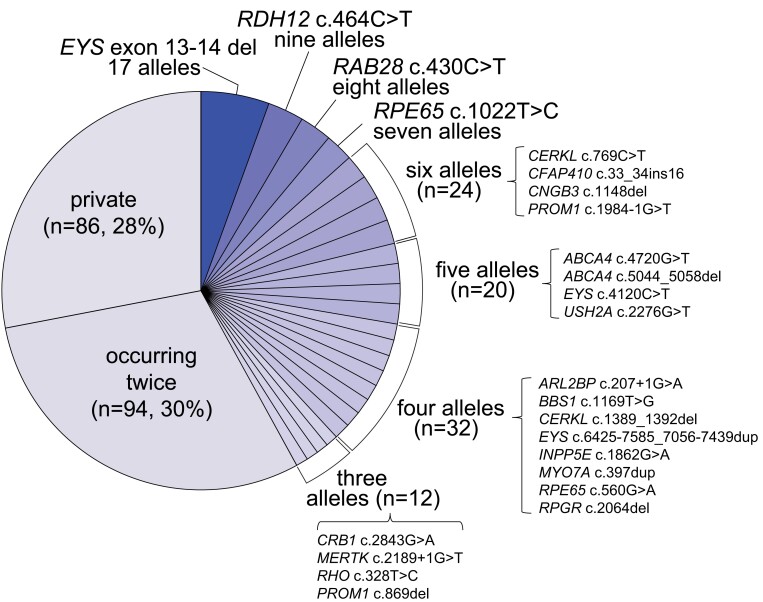
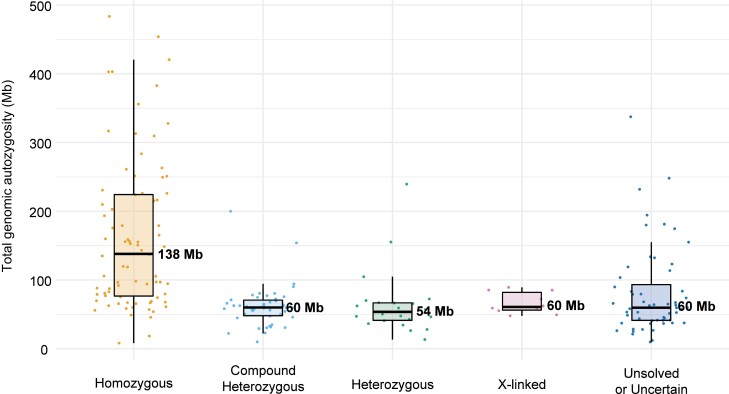
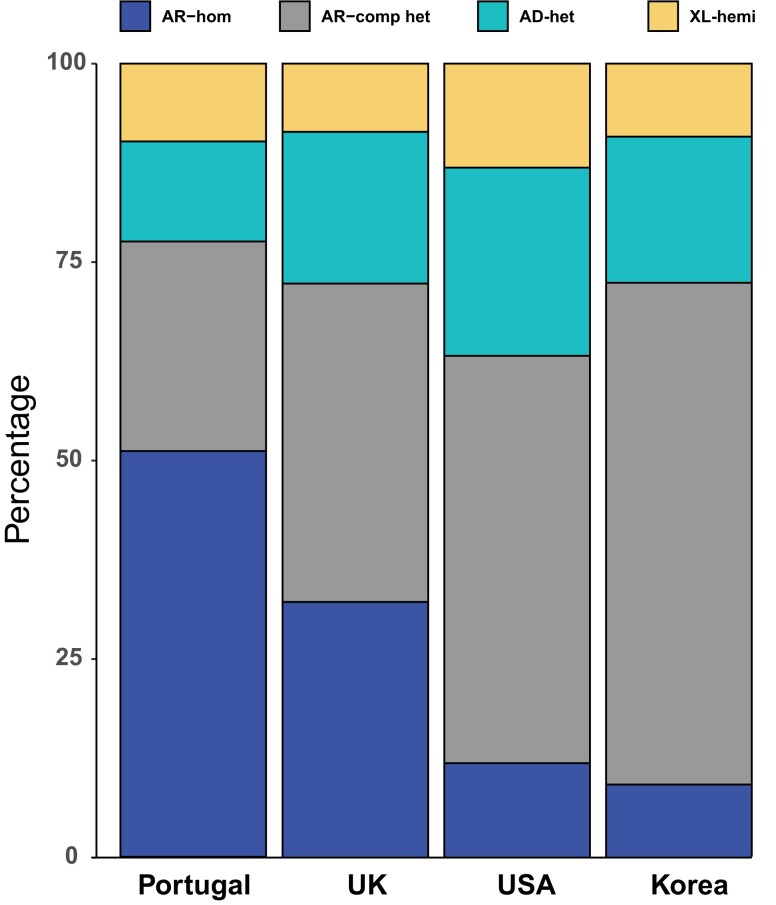
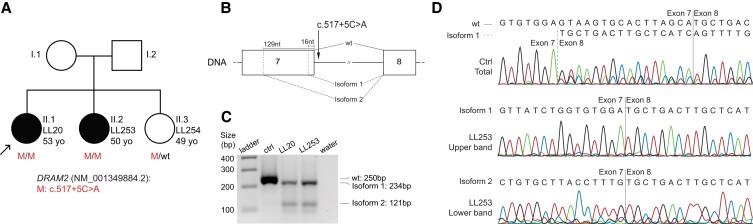
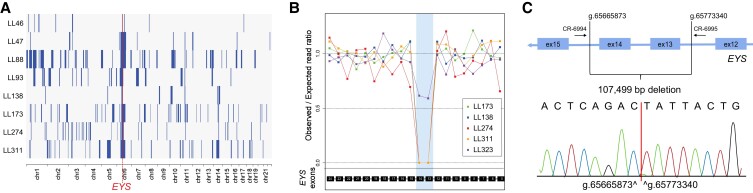
Inherited retinal diseases (IRDs) are a group of ocular conditions characterized by an elevated genetic and clinical heterogeneity. They are transmitted almost invariantly as monogenic traits. However, with more than 280 disease genes identified so far, association of clinical phenotypes with genotypes can be very challenging, and molecular diagnosis is essential for genetic counseling and correct management of the disease. In addition, the prevalence and the assortment of IRD mutations are often population-specific. In this work, we examined 230 families from Portugal, with individuals suffering from a variety of IRD diagnostic classes (270 subjects in total). Overall, we identified 157 unique mutations (34 previously unreported) in 57 distinct genes, with a diagnostic rate of 76%. The IRD mutational landscape was, to some extent, different from those reported in other European populations, including Spanish cohorts. For instance, the EYS gene appeared to be the most frequently mutated, with a prevalence of 10% among all IRD cases. This was, in part, due to the presence of a recurrent and seemingly founder mutation involving the deletion of exons 13 and 14 of this gene. Moreover, our analysis highlighted that as many as 51% of our cases had mutations in a homozygous state. To our knowledge, this is the first study assessing a cross-sectional genotype-phenotype landscape of IRDs in Portugal. Our data reveal a rather unique distribution of mutations, possibly shaped by a small number of rare ancestral events that have now become prevalent alleles in patients.
Keywords: EYS; Portugal; homozygosity; inherited retinal diseases.
© The Author(s) 2023. Published by Oxford University Press on behalf of National Academy of Sciences.
Figures
References
-
- Retinal Information Network (RetNet). https://web.sph.uth.edu/RetNet/.
-
- Ku CA, Pennesi ME. 2020. The new landscape of retinal gene therapy. Am J Med Genet C Semin Med Genet. 184:846–859. - PubMed
