

Polymerase θ inhibition activates the cGAS-STING pathway and cooperates with immune checkpoint blockade in models of BRCA-deficient cancer
- PMID: 36914658
- PMCID: PMC10011609
- DOI: 10.1038/s41467-023-37096-6
Polymerase θ inhibition activates the cGAS-STING pathway and cooperates with immune checkpoint blockade in models of BRCA-deficient cancer
Retraction in
-
Retraction Note: Polymerase θ inhibition activates the cGAS-STING pathway and cooperates with immune checkpoint blockade in models of BRCA-deficient cancer.Nat Commun. 2023 Dec 11;14(1):8193. doi: 10.1038/s41467-023-43803-0. Nat Commun. 2023. PMID: 38081854 Free PMC article. No abstract available.
Abstract
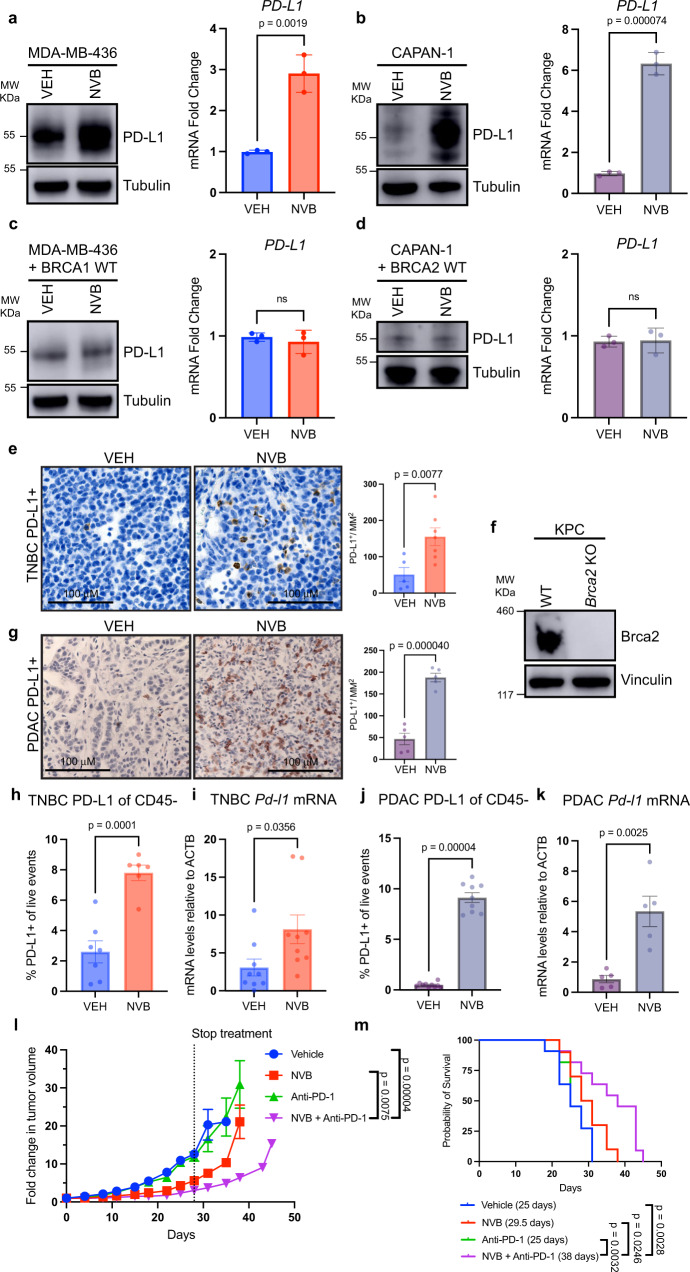
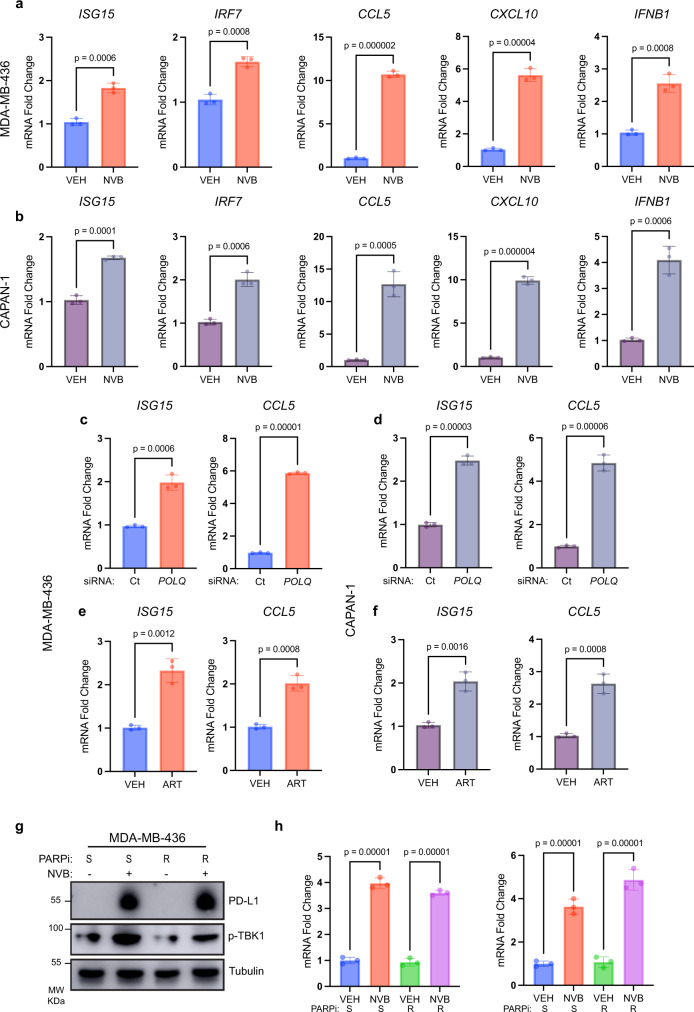
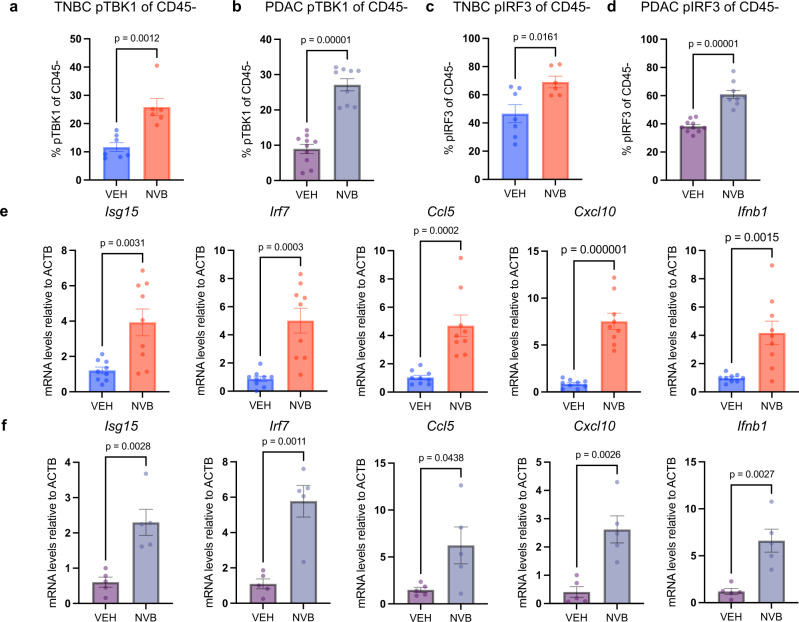
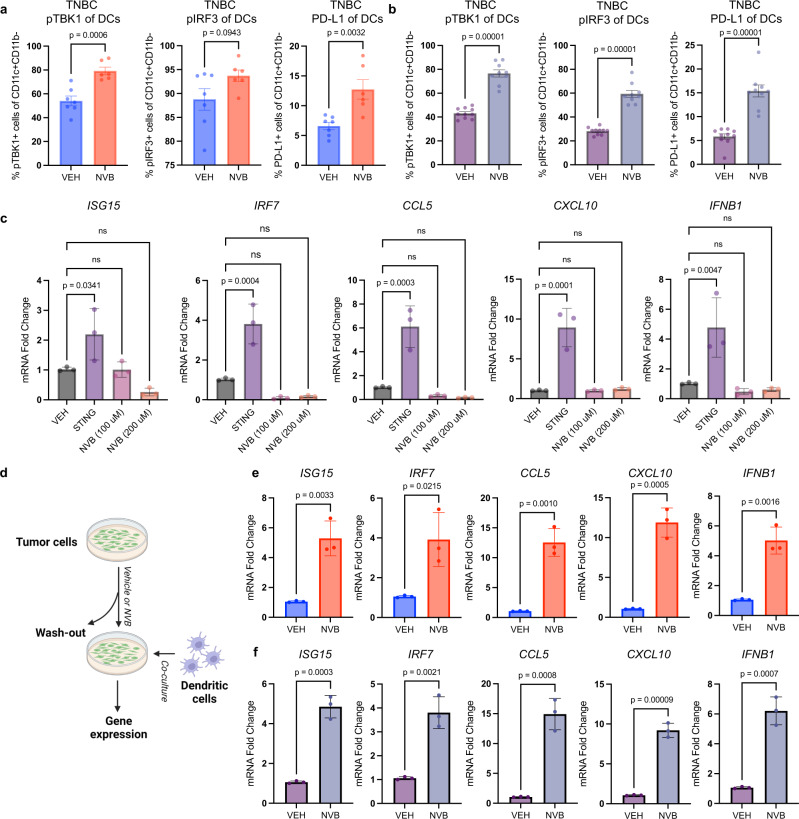
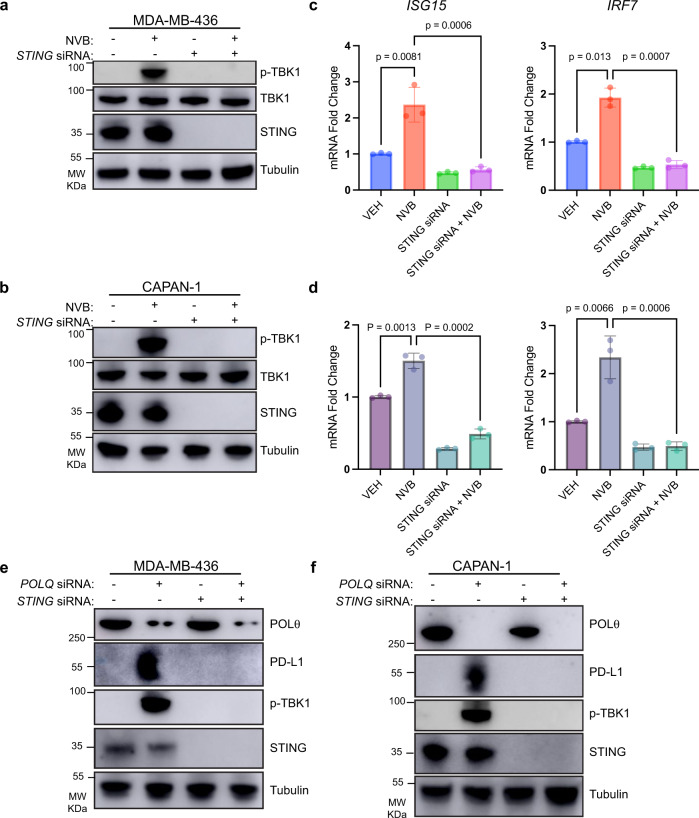
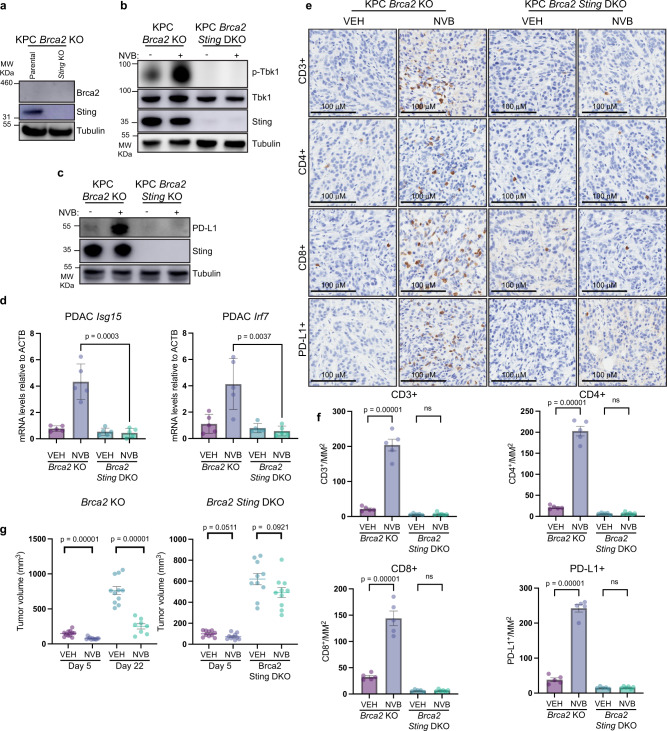
Recently developed inhibitors of polymerase theta (POLθ) have demonstrated synthetic lethality in BRCA-deficient tumor models. To examine the contribution of the immune microenvironment to antitumor efficacy, we characterized the effects of POLθ inhibition in immunocompetent models of BRCA1-deficient triple-negative breast cancer (TNBC) or BRCA2-deficient pancreatic ductal adenocarcinoma (PDAC). We demonstrate that genetic POLQ depletion or pharmacological POLθ inhibition induces both innate and adaptive immune responses in these models. POLθ inhibition resulted in increased micronuclei, cGAS/STING pathway activation, type I interferon gene expression, CD8+ T cell infiltration and activation, local paracrine activation of dendritic cells and upregulation of PD-L1 expression. Depletion of CD8+ T cells compromised the efficacy of POLθ inhibition, whereas antitumor effects were augmented in combination with anti-PD-1 immunotherapy. Collectively, our findings demonstrate that POLθ inhibition induces immune responses in a cGAS/STING-dependent manner and provide a rationale for combining POLθ inhibition with immune checkpoint blockade for the treatment of HR-deficient cancers.
© 2023. The Author(s).
Conflict of interest statement
J.L.G. is a consultant for Glaxo-Smith Kline (GSK), Codagenix, Verseau Therapeutics, Kymera, Kowa, Duke Street Bio., and Array BioPharma and receives sponsored research support from GSK, Array BioPharma, Merck and Eli Lilly. G.M.W. reports research funding from Merck & Co, institutional funding from Glaxo Smith Kline and Genentech, and US patent 20090258352, “A1 Pin1 as a marker for abnormal l cell growth,” licensed to Cell Signaling and R&D Systems. B.M.W. receives Research support from Celgene, Eli Lilly, Novartis, and Revolution Medicine; Consulting for Celgene, GRAIL, and Mirati. A.J.A. has consulted for Oncorus, Inc., Arrakis Therapeutics, Syros Pharmaceuticals, Boehringer Ingelheim, T-knife Therapeutics, AstraZeneca, Mirati Therapeutics, Revolution Medicines, Anji Pharmaceuticals, and Merck & Co., Inc. A.J.A. consults for and holds equity in Riva Therapeutics. A.J.A. has research funding from Mirati Therapeutics, Syros Pharmaceuticals, Bristol Myers Squibb, Revolution Medicines, Novartis, Novo Ventures and Deerfield, Inc. J.M.C. receives research funding to his institution from Merus, Roche, and Bristol Myers Squibb. He receives research support from Merck, AstraZeneca, Esperas Pharma, Bayer, Tesaro, Arcus Biosciences, and Apexigen; he received honoraria for being on advisory boards of Syros Pharmaceuticals, Incyte, and Blueprint Medicines. A.D.D. reports consulting for AstraZeneca, Bayer AG, Blacksmith/Lightstone Ventures, Bristol Myers Squibb, Cyteir Therapeutics, EMD Serono, Impact Therapeutics, PrimeFour Therapeutics, Pfizer, Tango Therapeutics, and Zentalis Pharmaceuticals/Zeno Management; is an Advisory Board member for Cyteir, and Impact Therapeutics; stockholder in Cedilla Therapeutics, Cyteir, Impact Therapeutics, and PrimeFour Therapeutics; and reports receiving commercial research grants from Bristol Myers Squibb, EMD Serono, Moderna, and Tango Therapeutics. G.I.S. has received research funding from Eli Lilly, Merck KGaA/EMD-Serono, Merck & Co., and Pfizer. He has served on advisory boards for Pfizer, Eli Lilly, Merck KGaA/EMD-Serono, Bicycle Therapeutics, Fusion Pharmaceuticals, Cybrexa Therapeutics, Bayer, Boehringer Ingelheim, ImmunoMet, Artios, Atrin, Concarlo Holdings, Syros, Zentalis, CytomX Therapeutics, Blueprint Medicines, Kymera Therapeutics, Janssen and Xinthera. In addition, he holds a patent entitled, “Dosage regimen for sapacitabine and seliciclib,” also issued to Cyclacel Pharmaceuticals, and a pending patent, entitled, “Compositions and Methods for Predicting Response and Resistance to CDK4/6 Inhibition,” together with Liam Cornell. The remaining authors declare no competing interests.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
