

The P323L substitution in the SARS-CoV-2 polymerase (NSP12) confers a selective advantage during infection
- PMID: 36915185
- PMCID: PMC10009825
- DOI: 10.1186/s13059-023-02881-5
The P323L substitution in the SARS-CoV-2 polymerase (NSP12) confers a selective advantage during infection
Abstract
Background: The mutational landscape of SARS-CoV-2 varies at the dominant viral genome sequence and minor genomic variant population. During the COVID-19 pandemic, an early substitution in the genome was the D614G change in the spike protein, associated with an increase in transmissibility. Genomes with D614G are accompanied by a P323L substitution in the viral polymerase (NSP12). However, P323L is not thought to be under strong selective pressure.
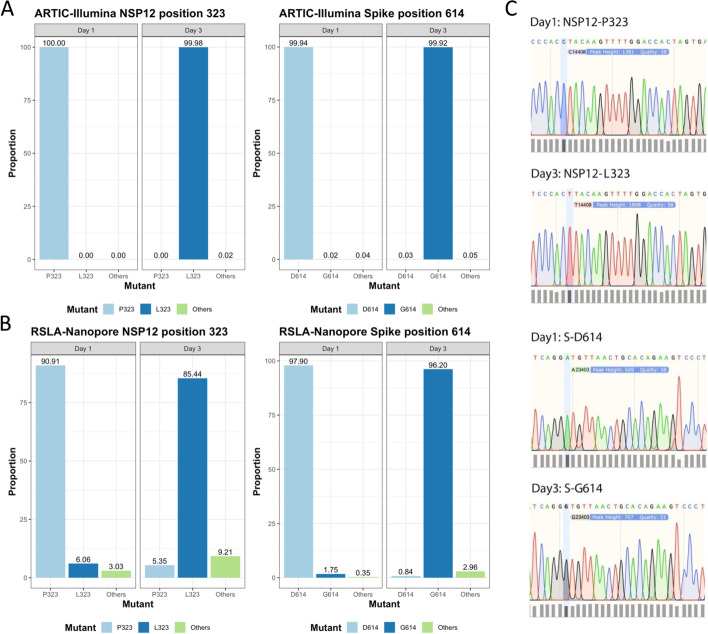
Results: Investigation of P323L/D614G substitutions in the population shows rapid emergence during the containment phase and early surge phase during the first wave. These substitutions emerge from minor genomic variants which become dominant viral genome sequence. This is investigated in vivo and in vitro using SARS-CoV-2 with P323 and D614 in the dominant genome sequence and L323 and G614 in the minor variant population. During infection, there is rapid selection of L323 into the dominant viral genome sequence but not G614. Reverse genetics is used to create two viruses (either P323 or L323) with the same genetic background. L323 shows greater abundance of viral RNA and proteins and a smaller plaque morphology than P323.
Conclusions: These data suggest that P323L is an important contribution in the emergence of variants with transmission advantages. Sequence analysis of viral populations suggests it may be possible to predict the emergence of a new variant based on tracking the frequency of minor variant genomes. The ability to predict an emerging variant of SARS-CoV-2 in the global landscape may aid in the evaluation of medical countermeasures and non-pharmaceutical interventions.
Keywords: COVID-19; Evolution; NSP12; P323L; Polymerase; SARS-CoV-2; Selection; Spike protein.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures






References
-
- Davidson AD, Williamson MK, Lewis S, Shoemark D, Carroll MW, Heesom KJ, Zambon M, Ellis J, Lewis PA, Hiscox JA, Matthews DA. Characterisation of the transcriptome and proteome of SARS-CoV-2 reveals a cell passage induced in-frame deletion of the furin-like cleavage site from the spike glycoprotein. Genome Med. 2020;12:68. doi: 10.1186/s13073-020-00763-0. - DOI - PMC - PubMed
-
- Young BE, Fong SW, Chan YH, Mak TM, Ang LW, Anderson DE, Lee CY, Amrun SN, Lee B, Goh YS, et al. Effects of a major deletion in the SARS-CoV-2 genome on the severity of infection and the inflammatory response: an observational cohort study. Lancet. 2020;396:603–611. doi: 10.1016/S0140-6736(20)31757-8. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
