

Reassessment of weak parent-of-origin expression bias shows it rarely exists outside of known imprinted regions
- PMID: 36916665
- PMCID: PMC10097420
- DOI: 10.7554/eLife.83364
Reassessment of weak parent-of-origin expression bias shows it rarely exists outside of known imprinted regions
Abstract
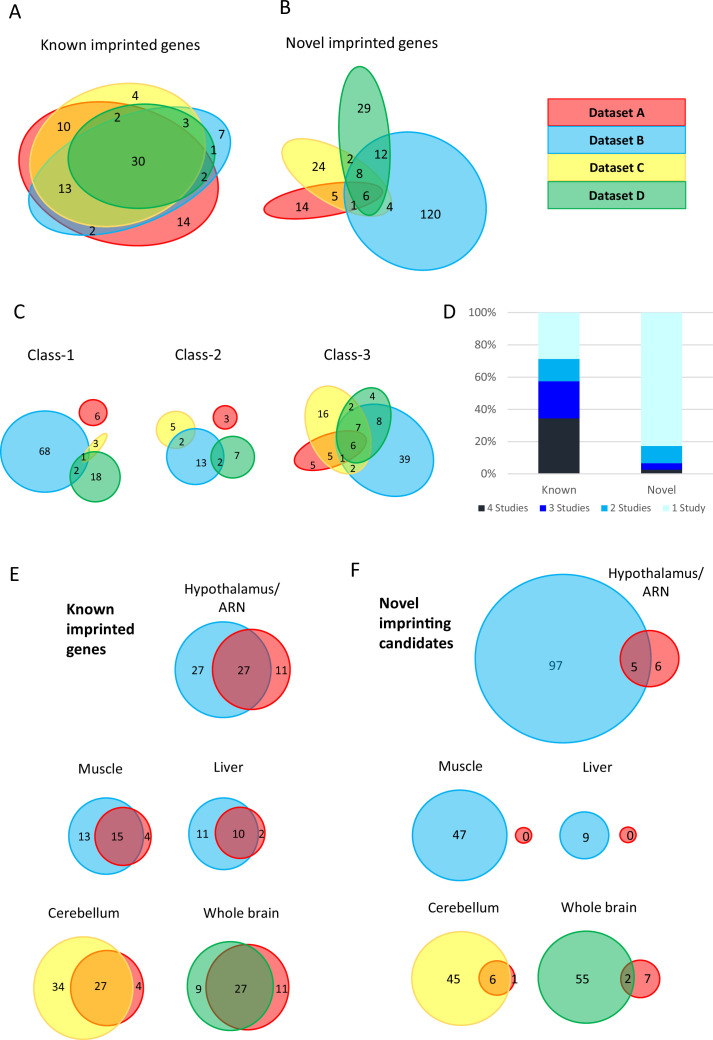
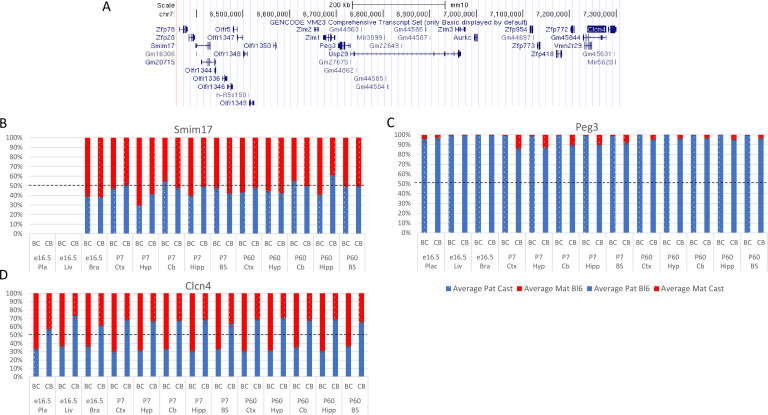
In mouse and human, genes subjected to genomic imprinting have been shown to function in development, behavior, and post-natal adaptations. Failure to correctly imprint genes in human is associated with developmental syndromes, adaptive, and metabolic disorders during life as well as numerous forms of cancer. In recent years researchers have turned to RNA-seq technologies applied to reciprocal hybrid strains of mice to identify novel imprinted genes, causing a threefold increase in genes reported as having a parental origin-specific expression bias. The functional relevance of parental origin-specific expression bias is not fully appreciated especially since many are reported with only minimal parental bias (e.g. 51:49). Here, we present an in-depth meta-analysis of previously generated RNA-seq data and show that the methods used to generate and analyze libraries greatly influence the calling of allele-specific expression. Validation experiments show that most novel genes called with parental-origin-specific allelic bias are artefactual, with the mouse strain contributing a larger effect on expression biases than parental origin. Of the weak novel genes that do validate, most are located at the periphery of known imprinted domains, suggesting they may be affected by local allele- and tissue-specific conformation. Together these findings highlight the need for robust tools, definitions, and validation of putative imprinted genes to provide meaningful information within imprinting databases and to understand the functional and mechanistic implications of the process.
Keywords: epigenetics; gene expression; genetics; genomics; imprinting; mouse.
© 2023, Edwards et al.
Conflict of interest statement
CE, WW, ST, LH, RH, AF No competing interests declared
Figures













Update of
- doi: 10.1101/2022.08.21.504536
References
-
- Andergassen D, Dotter CP, Wenzel D, Sigl V, Bammer PC, Muckenhuber M, Mayer D, Kulinski TM, Theussl H-C, Penninger JM, Bock C, Barlow DP, Pauler FM, Hudson QJ. Mapping the mouse Allelome reveals tissue-specific regulation of allelic expression. eLife. 2017;6:e25125. doi: 10.7554/eLife.25125. - DOI - PMC - PubMed
Publication types
MeSH terms
Associated data
- SRA/SRP020526
- SRA/SRP056236
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
