

A homozygous POLR1A variant causes leukodystrophy and affects protein homeostasis
- PMID: 36917474
- PMCID: PMC10393412
- DOI: 10.1093/brain/awad086
A homozygous POLR1A variant causes leukodystrophy and affects protein homeostasis
Abstract
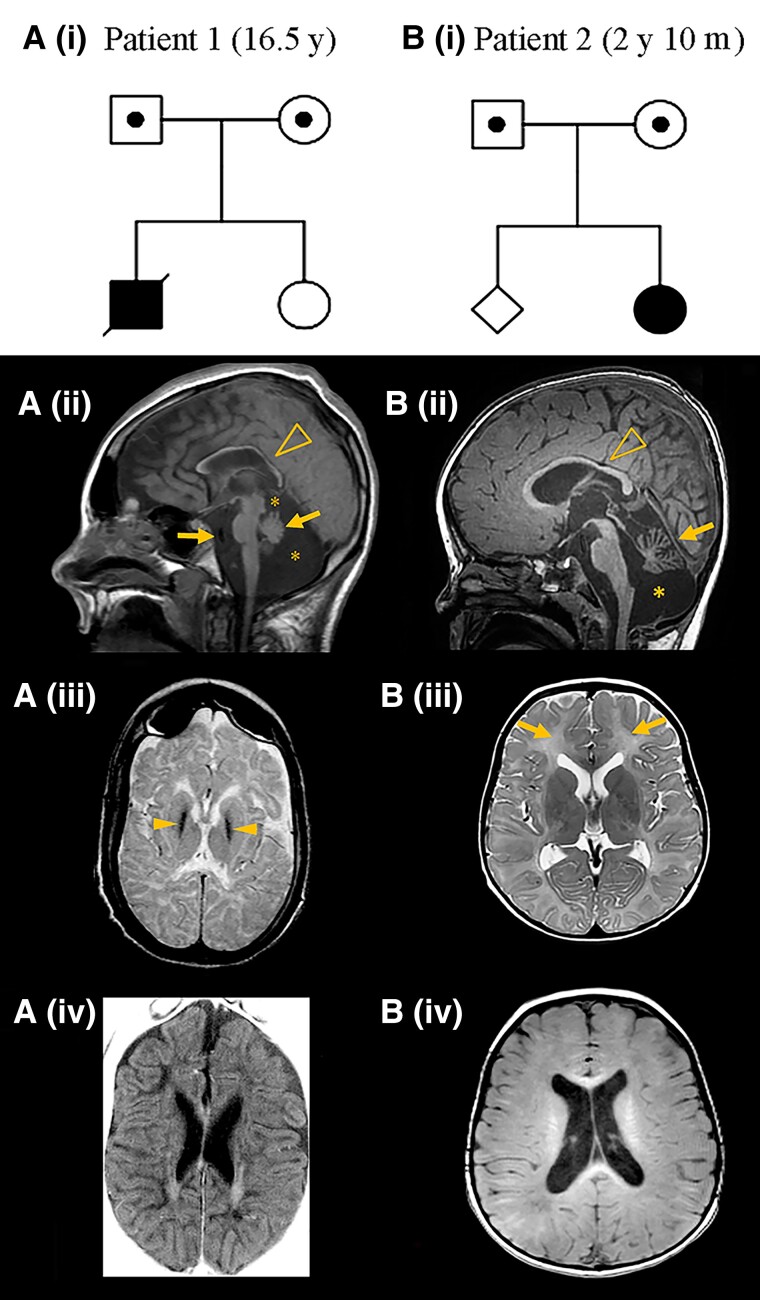
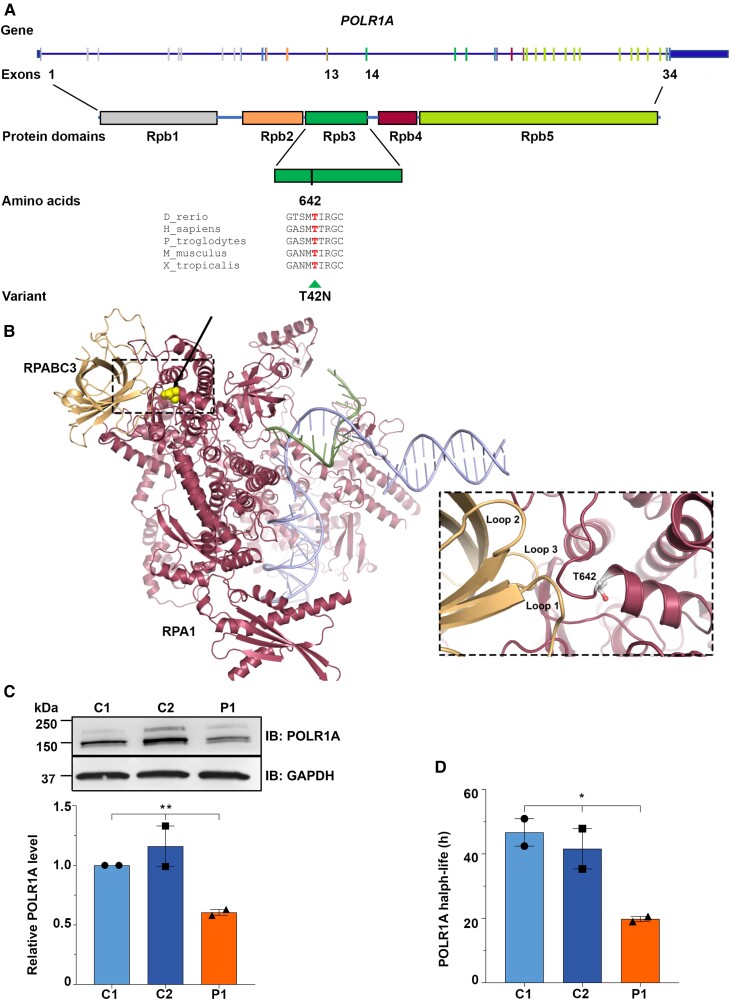
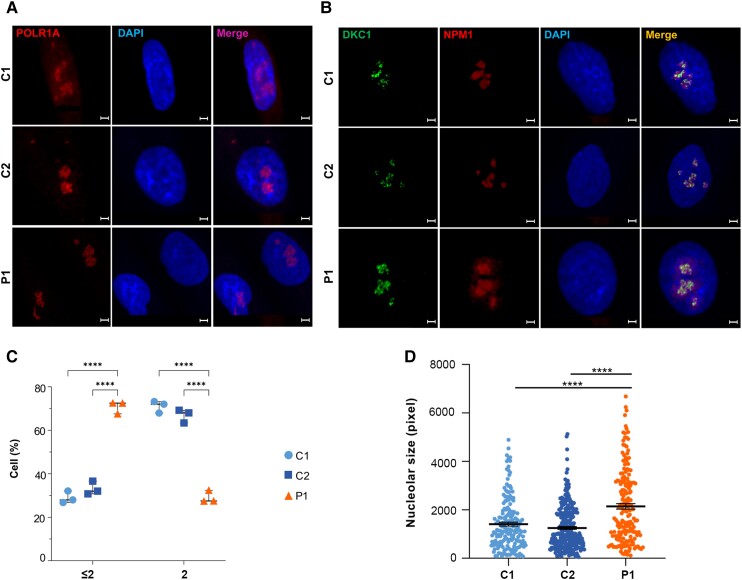
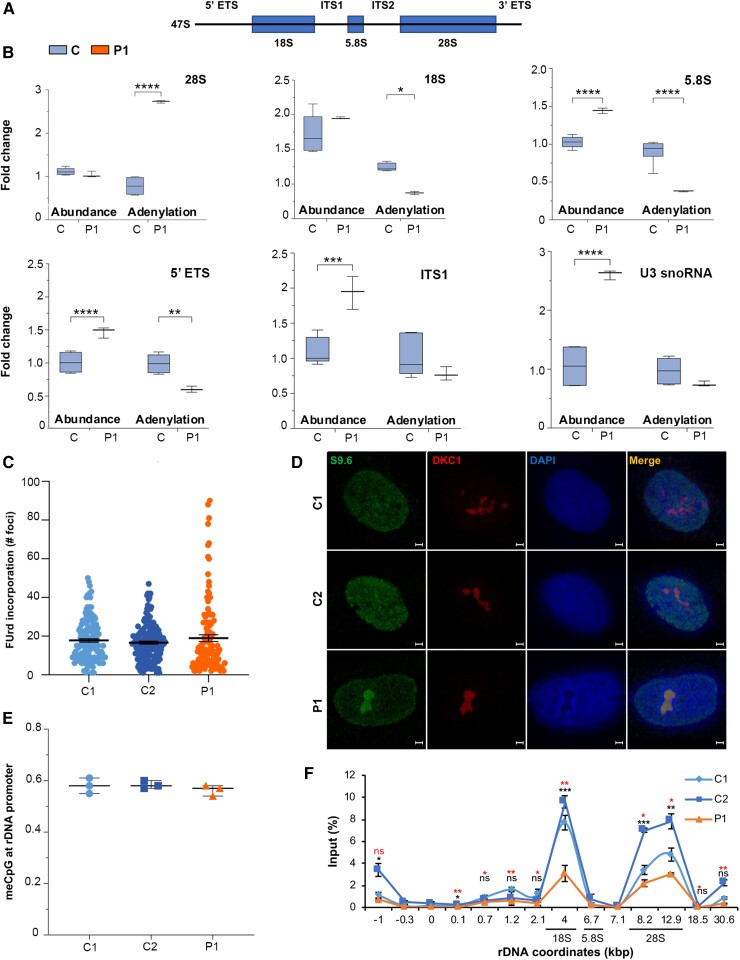
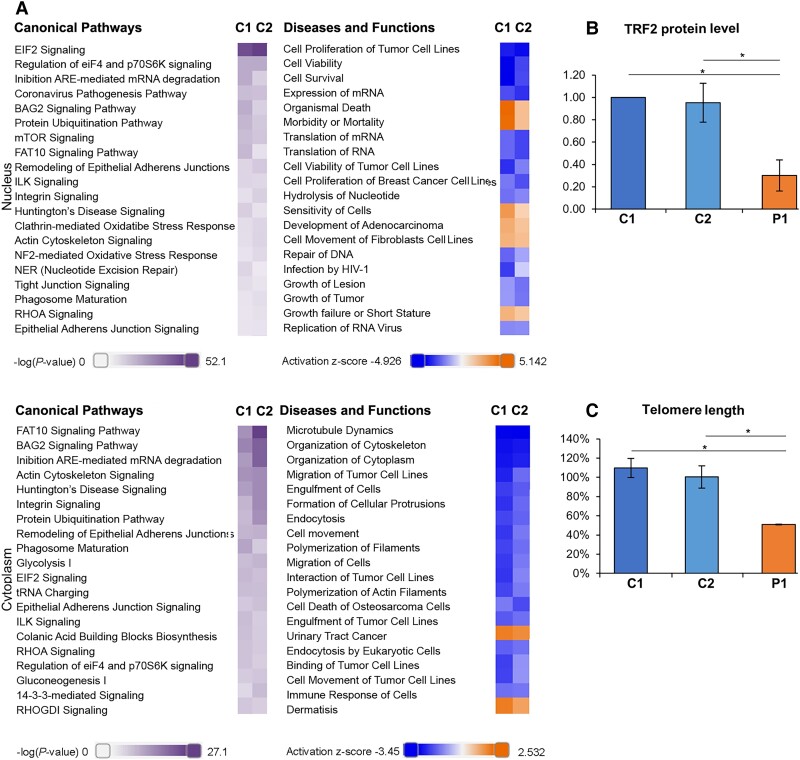
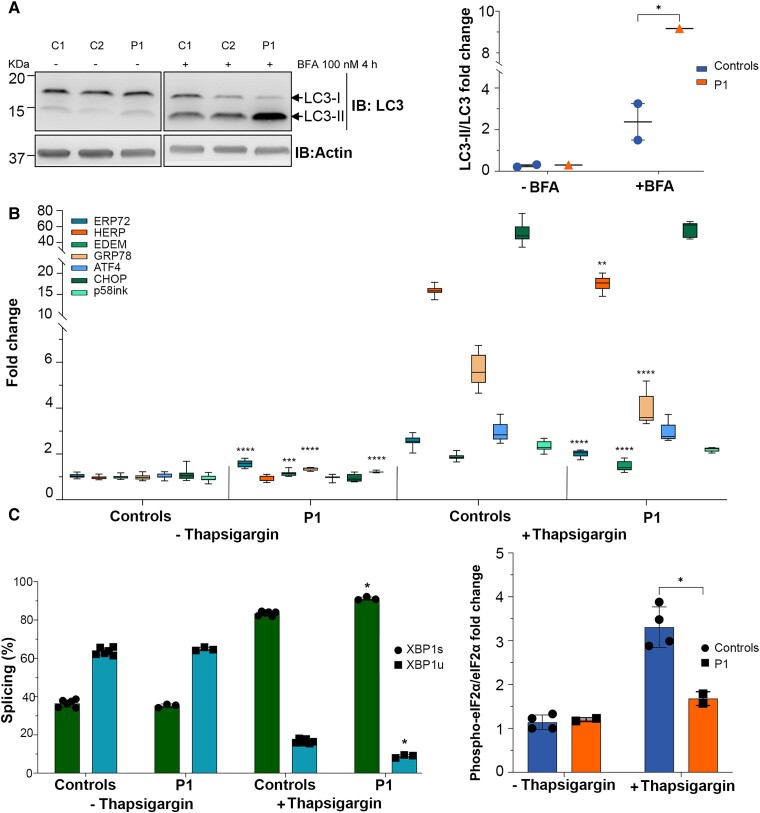
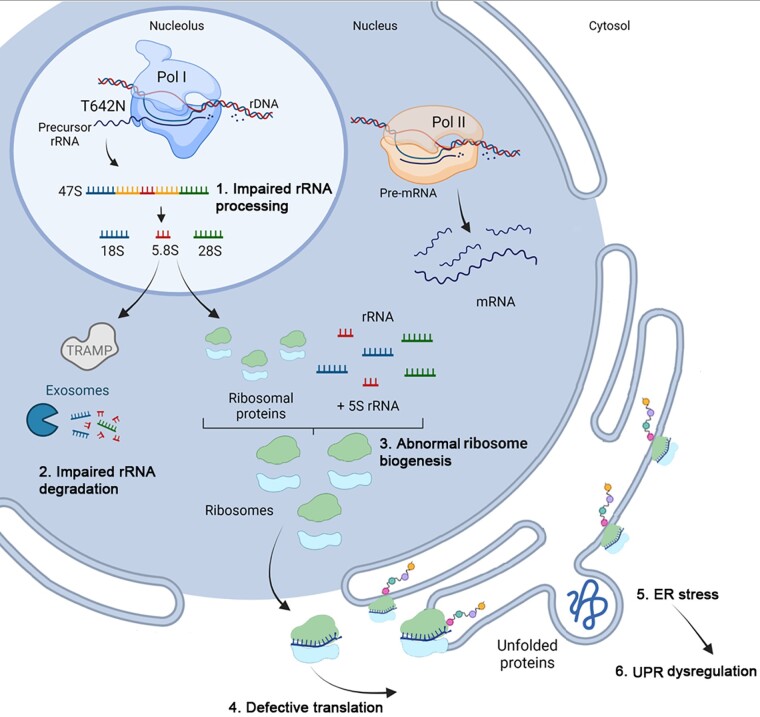
RNA polymerase I transcribes ribosomal DNA to produce precursor 47S rRNA. Post-transcriptional processing of this rRNA generates mature 28S, 18S and 5.8S rRNAs, which form the ribosomes, together with 5S rRNA, assembly factors and ribosomal proteins. We previously reported a homozygous variant in the catalytic subunit of RNA polymerase I, POLR1A, in two brothers with leukodystrophy and progressive course. However, the disease mechanism remained unknown. In this report, we describe another missense variant POLR1A NM_015425.3:c.1925C>A; p.(Thr642Asn) in homozygosity in two unrelated patients. Patient 1 was a 16-year-old male and Patient 2 was a 2-year-old female. Both patients manifested neurological deficits, with brain MRIs showing hypomyelinating leukodystrophy and cerebellar atrophy; and in Patient 1 additionally with hypointensity of globi pallidi and small volume of the basal ganglia. Patient 1 had progressive disease course, leading to death at the age of 16.5 years. Extensive in vitro experiments in fibroblasts from Patient 1 documented that the mutated POLR1A led to aberrant rRNA processing and degradation, and abnormal nucleolar homeostasis. Proteomics data analyses and further in vitro experiments documented abnormal protein homeostasis, and endoplasmic reticulum stress responses. We confirm that POLR1A biallelic variants cause neurodegenerative disease, expand the knowledge of the clinical phenotype of the disorder, and provide evidence for possible pathological mechanisms leading to POLR1A-related leukodystrophy.
Keywords: POLR1A; myelin; neurodegeneration; rRNA; ribosome.
© The Author(s) 2023. Published by Oxford University Press on behalf of the Guarantors of Brain.
Conflict of interest statement
The authors report no competing interests.
Figures
References
-
- Shenoy RD, Shetty V, Dheedene A, et al. Phenotypic and molecular heterogeneity in mandibulofacial dysostoses: A case series from India. Cleft Palate Craniofac J. 2021;59:1346–1351. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
