

Gasdermin-E mediates mitochondrial damage in axons and neurodegeneration
- PMID: 36917977
- PMCID: PMC10121894
- DOI: 10.1016/j.neuron.2023.02.019
Gasdermin-E mediates mitochondrial damage in axons and neurodegeneration
Abstract
Mitochondrial dysfunction and axon loss are hallmarks of neurologic diseases. Gasdermin (GSDM) proteins are executioner pore-forming molecules that mediate cell death, yet their roles in the central nervous system (CNS) are not well understood. Here, we find that one GSDM family member, GSDME, is expressed by both mouse and human neurons. GSDME plays a role in mitochondrial damage and axon loss. Mitochondrial neurotoxins induced caspase-dependent GSDME cleavage and rapid localization to mitochondria in axons, where GSDME promoted mitochondrial depolarization, trafficking defects, and neurite retraction. Frontotemporal dementia (FTD)/amyotrophic lateral sclerosis (ALS)-associated proteins TDP-43 and PR-50 induced GSDME-mediated damage to mitochondria and neurite loss. GSDME knockdown protected against neurite loss in ALS patient iPSC-derived motor neurons. Knockout of GSDME in SOD1G93A ALS mice prolonged survival, ameliorated motor dysfunction, rescued motor neuron loss, and reduced neuroinflammation. We identify GSDME as an executioner of neuronal mitochondrial dysfunction that may contribute to neurodegeneration.
Keywords: ALS; FTD; axon degeneration; cell death; gasdermins; innate immunity; mitochondria; neurodegeneration; neuroimmunology; pyroptosis.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests I.M.C. receives sponsored research support from Abbvie/Allergan Pharmaceuticals and is on the SAB for GSK and LIMM therapeutics. J.L. is a cofounder and SAB member of Ventus Therapeutics.
Figures
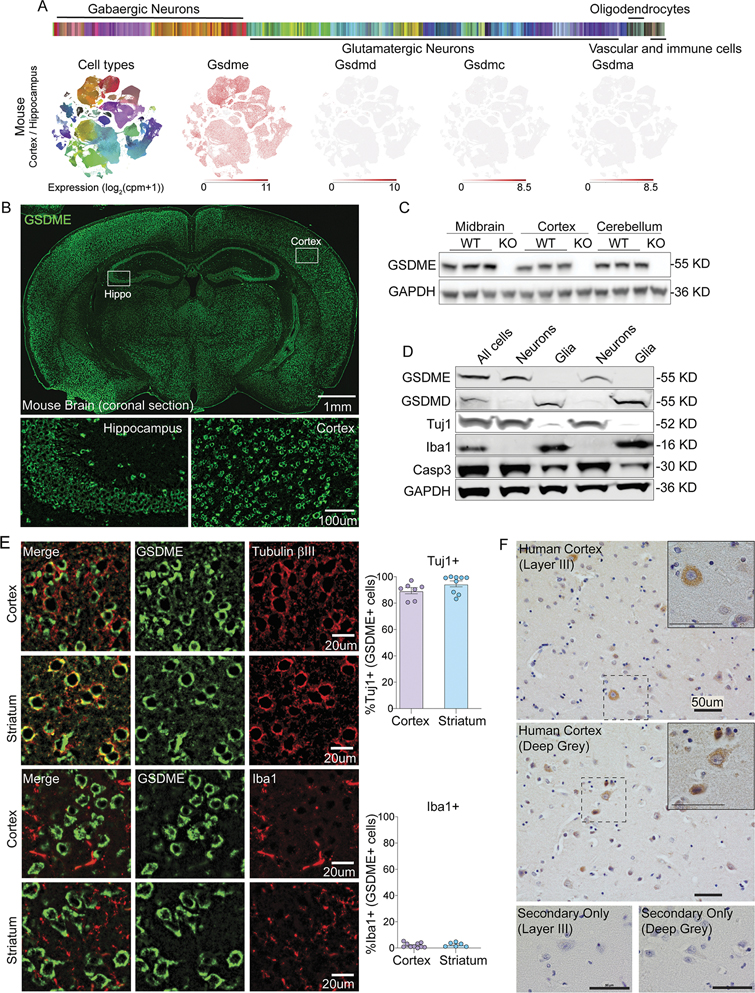
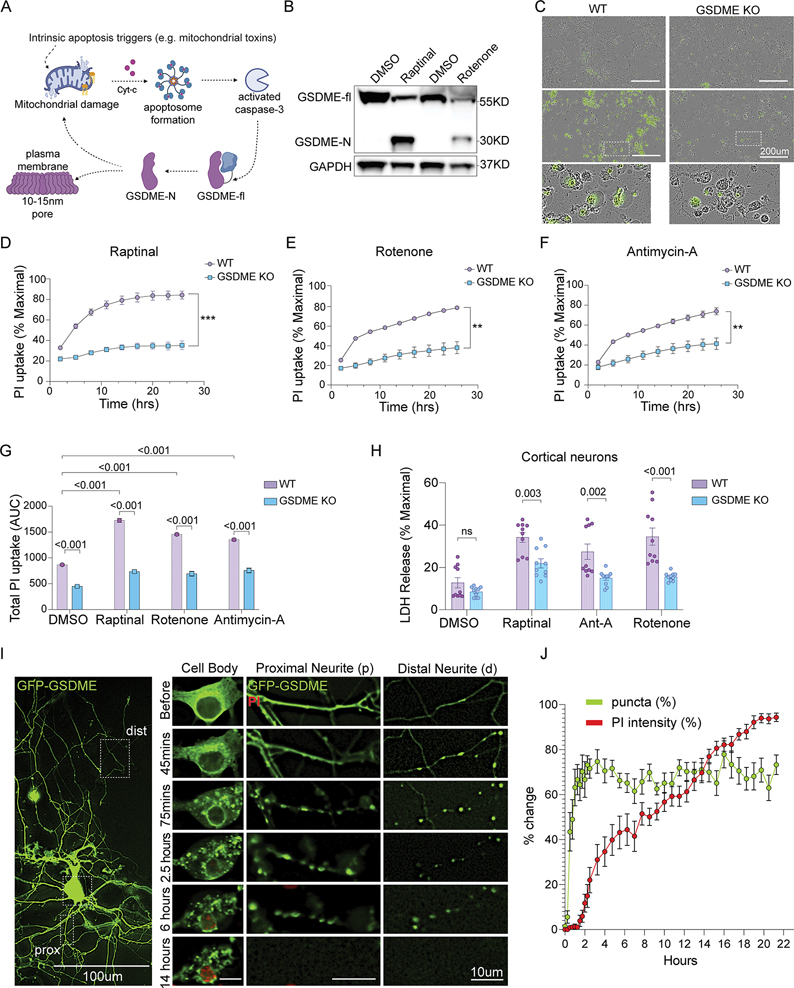
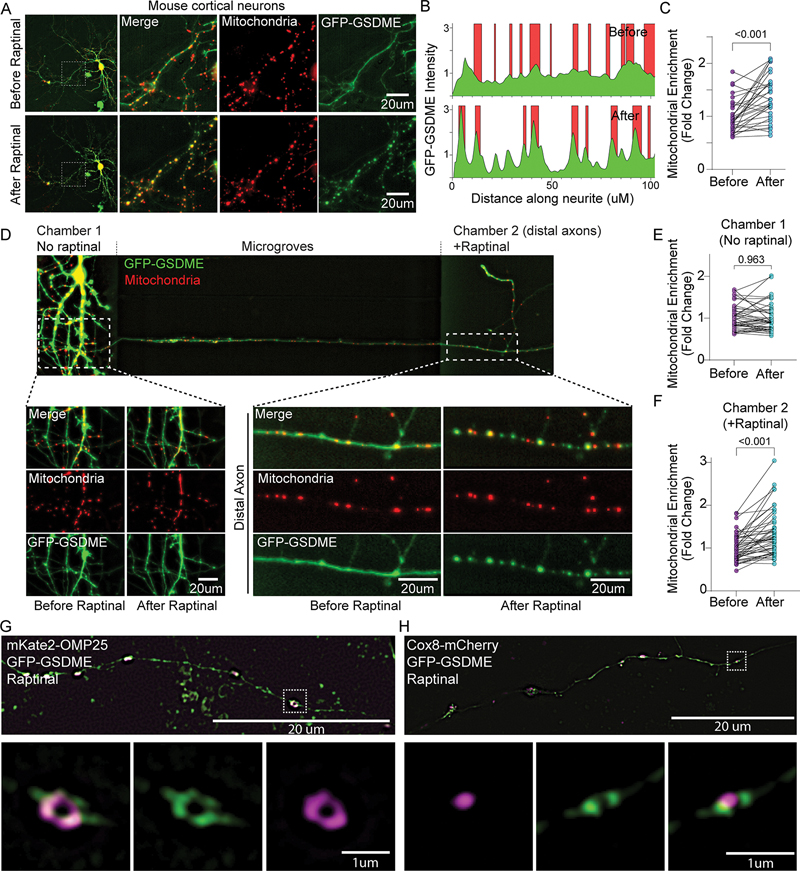
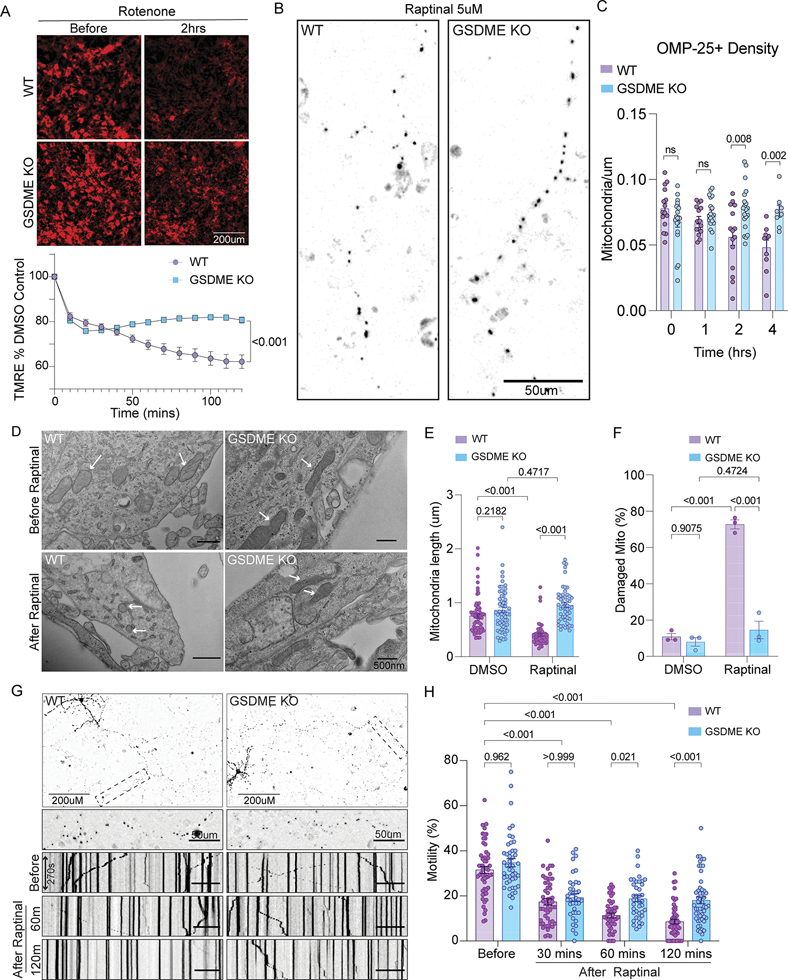
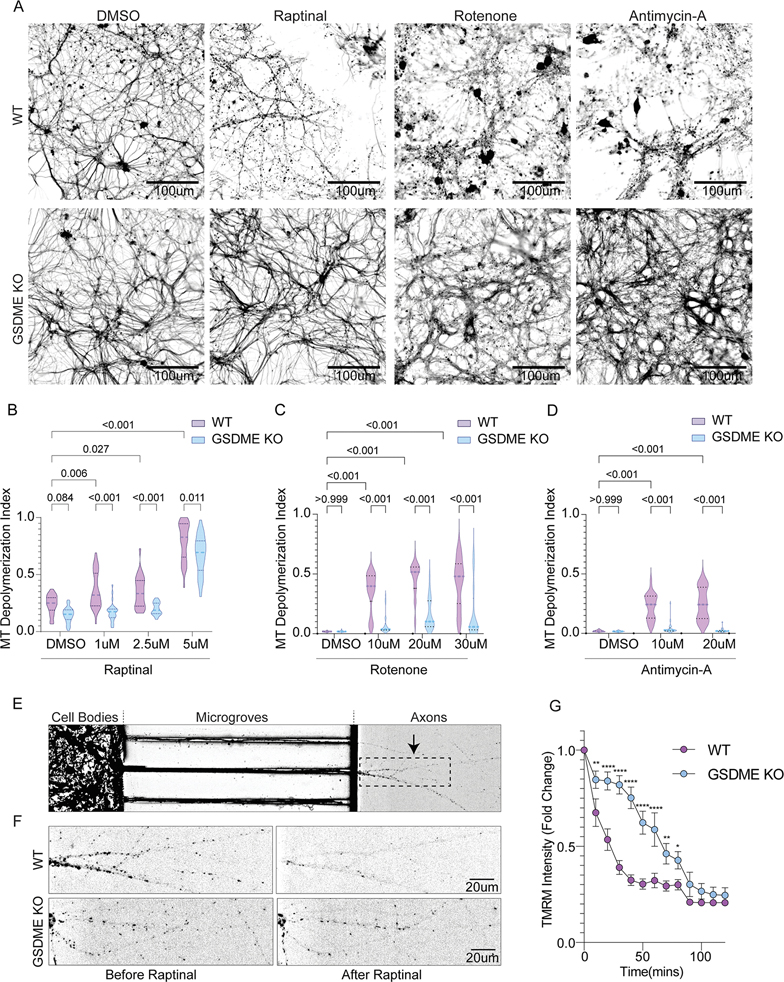
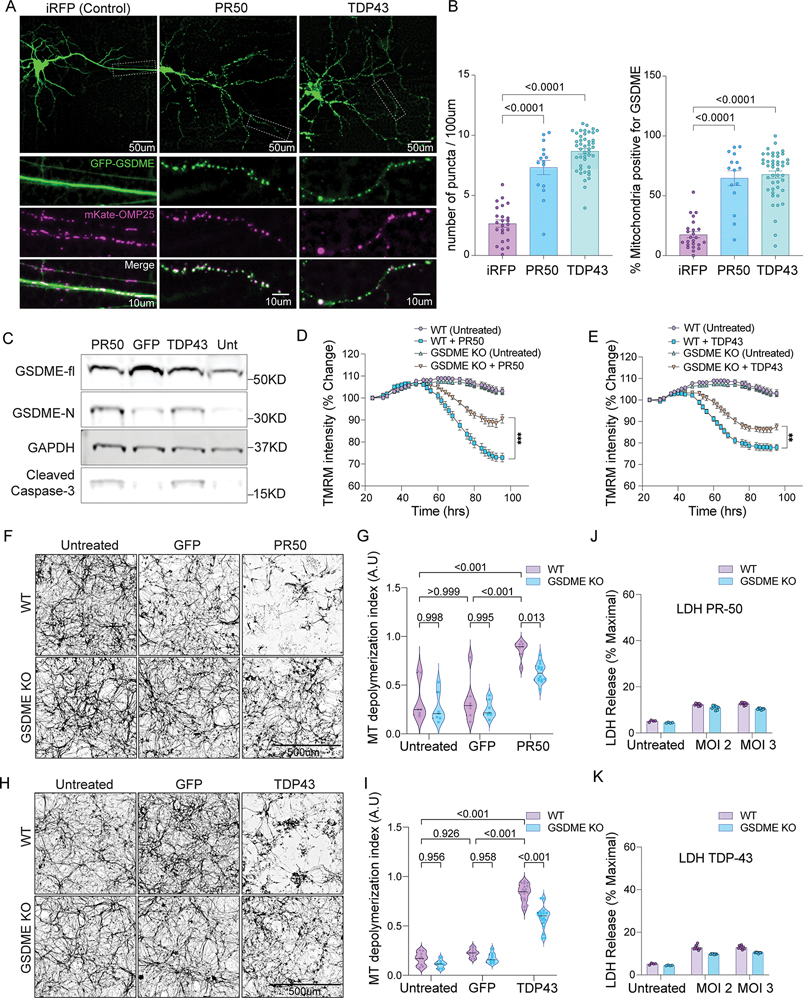
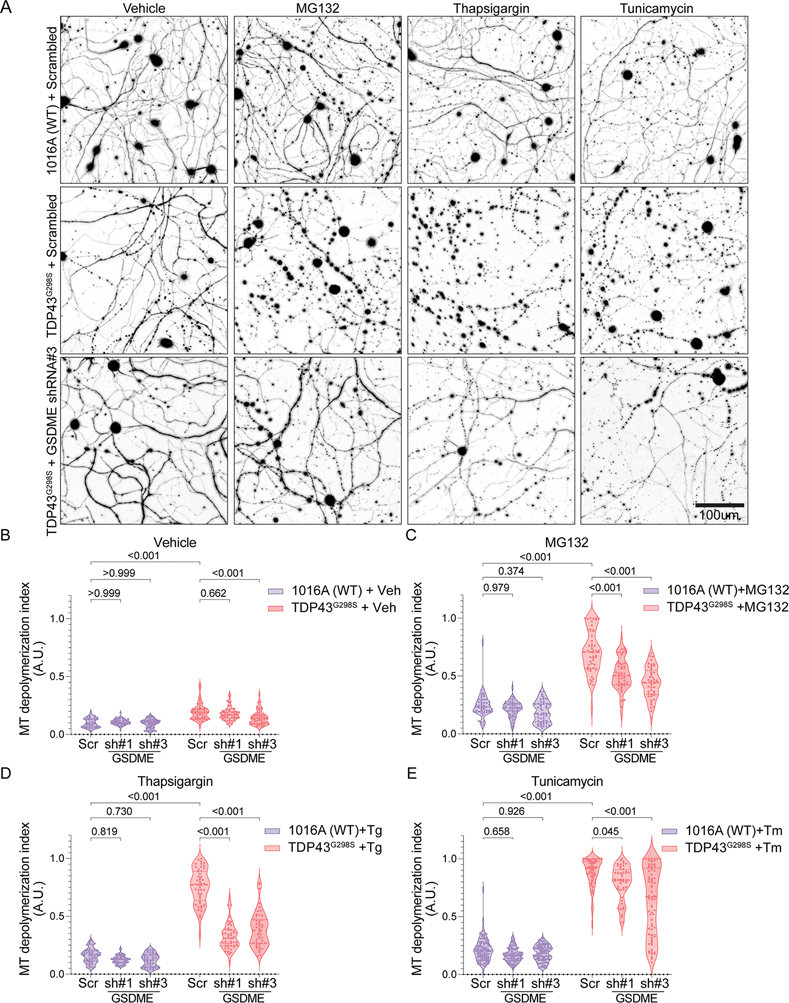
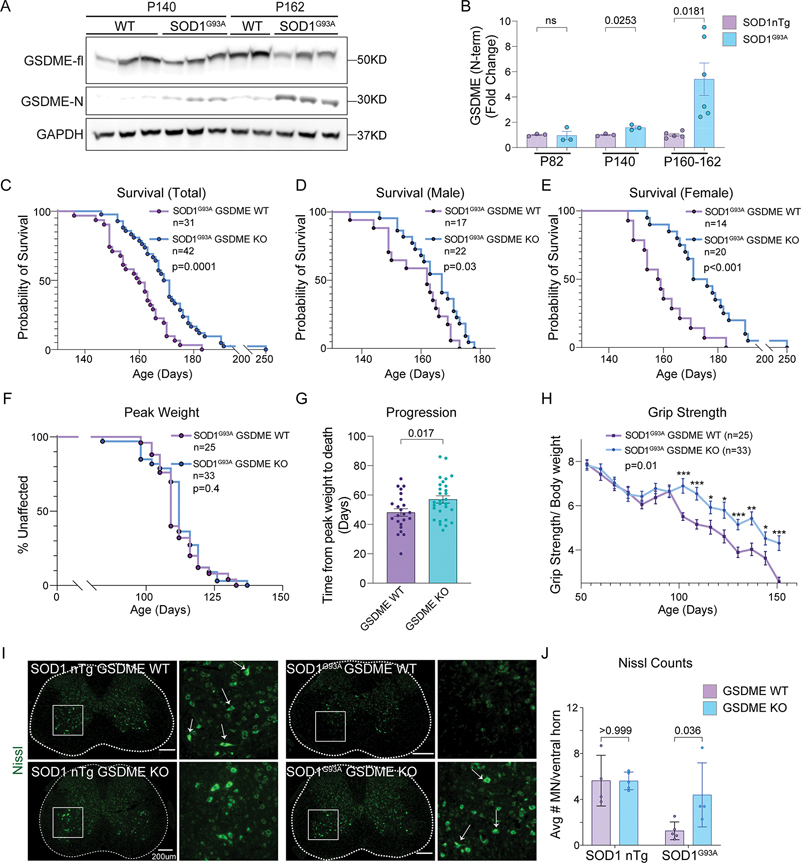
References
-
- Fuentes L, Lebenkoff S, White K, Gerdts C, Hopkins K, Potter JE, Grossman D, Project PE, and Sciences R (2016). Releasing Syntaphilin Removes Stressed Mitochondria from Axons Independent of Mitophagy under Pathophysiological Conditions. Neuron 93, 292–297. 10.1016/j.neuron.2017.04.004.Releasing. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
