

Sequence and chromatin features guide DNA double-strand break resection initiation
- PMID: 36917982
- PMCID: PMC10131398
- DOI: 10.1016/j.molcel.2023.02.010
Sequence and chromatin features guide DNA double-strand break resection initiation
Abstract
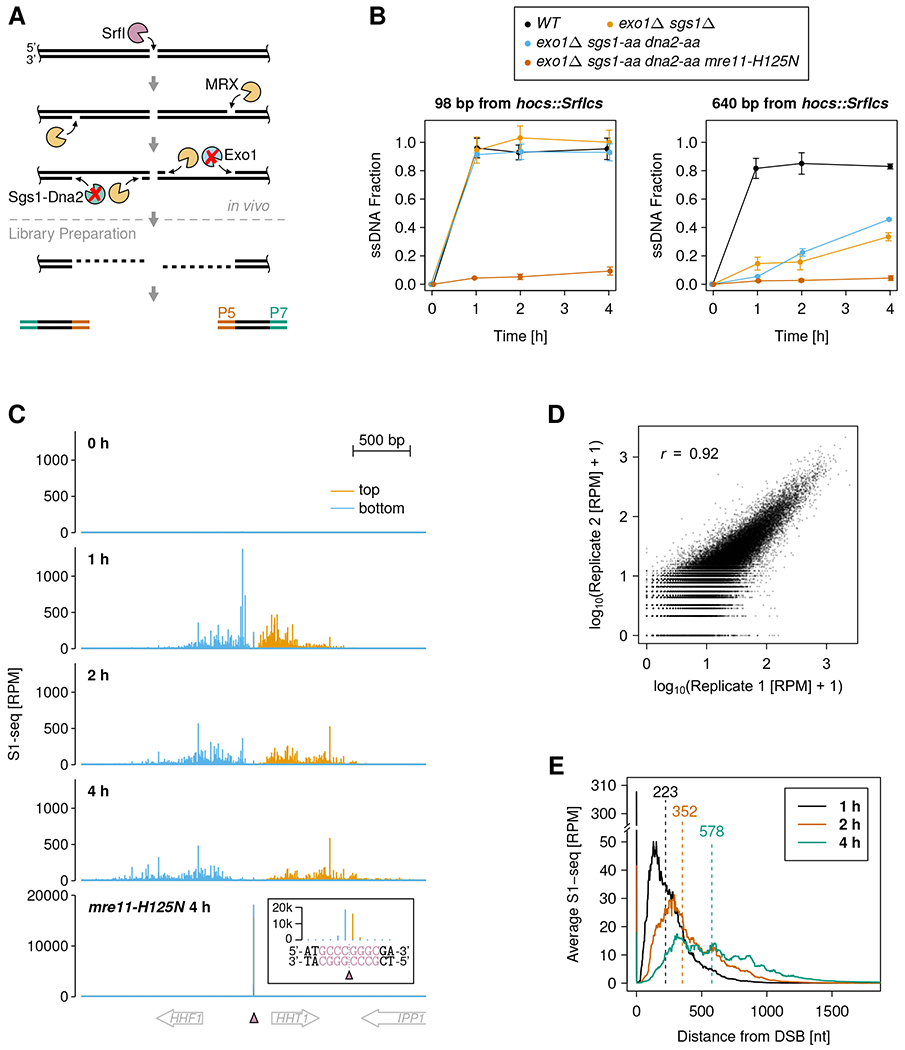
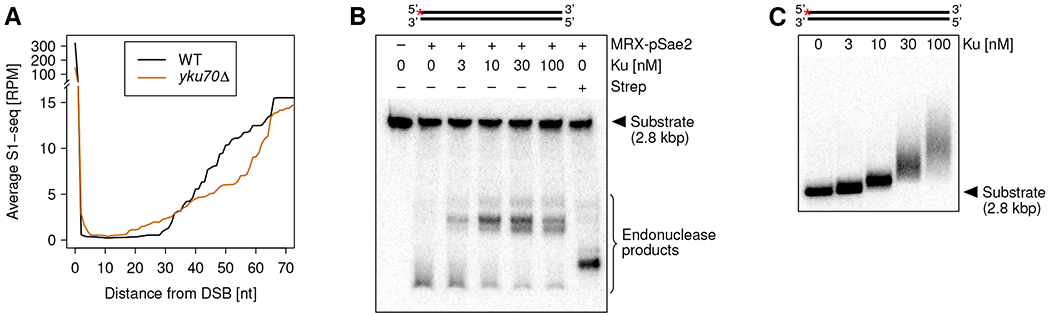
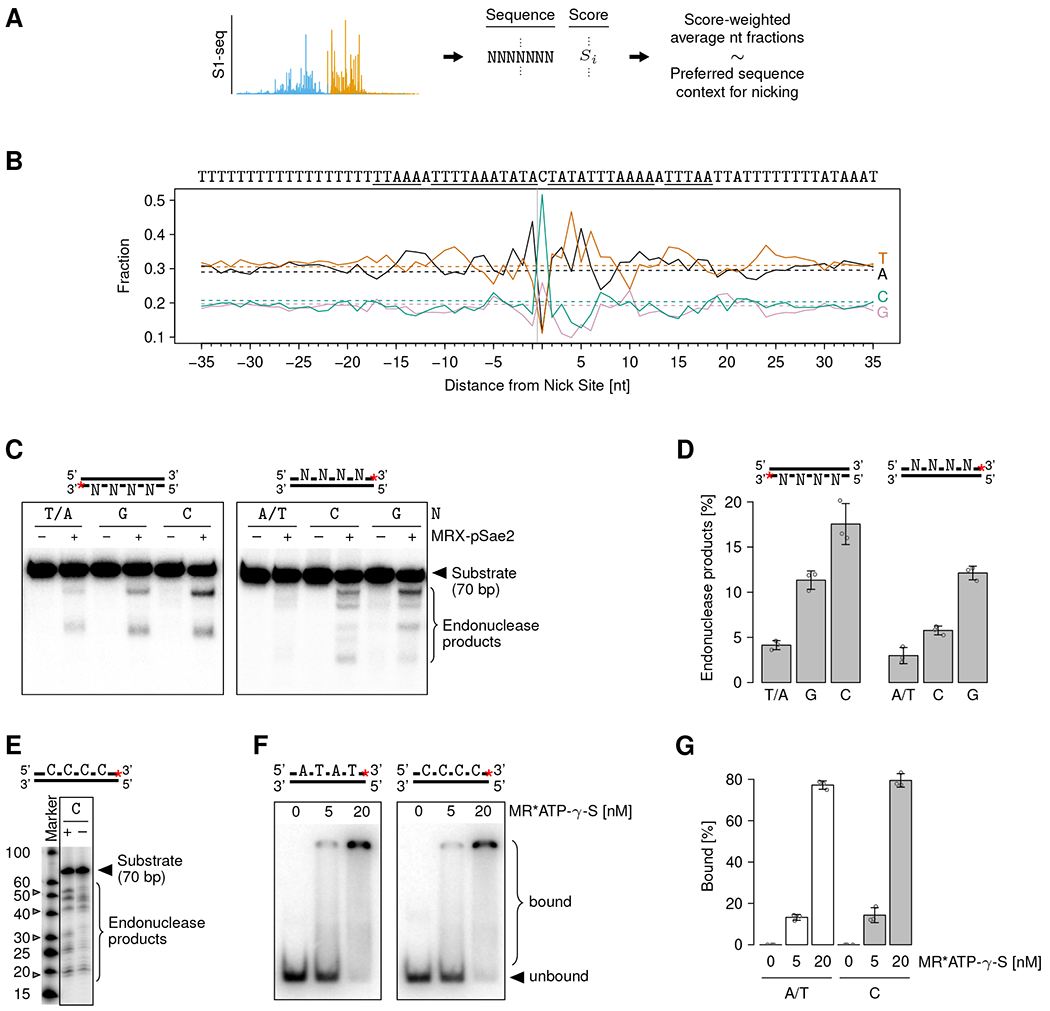
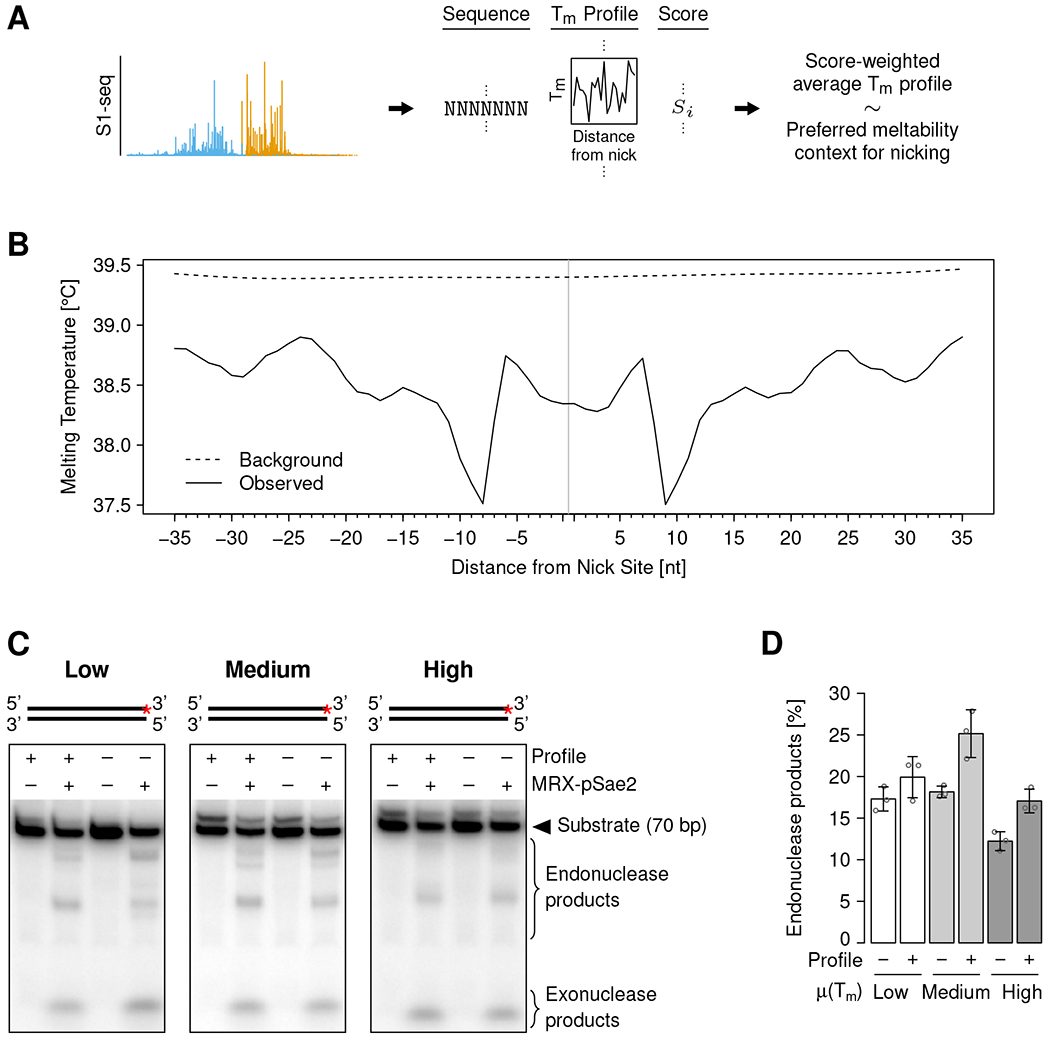
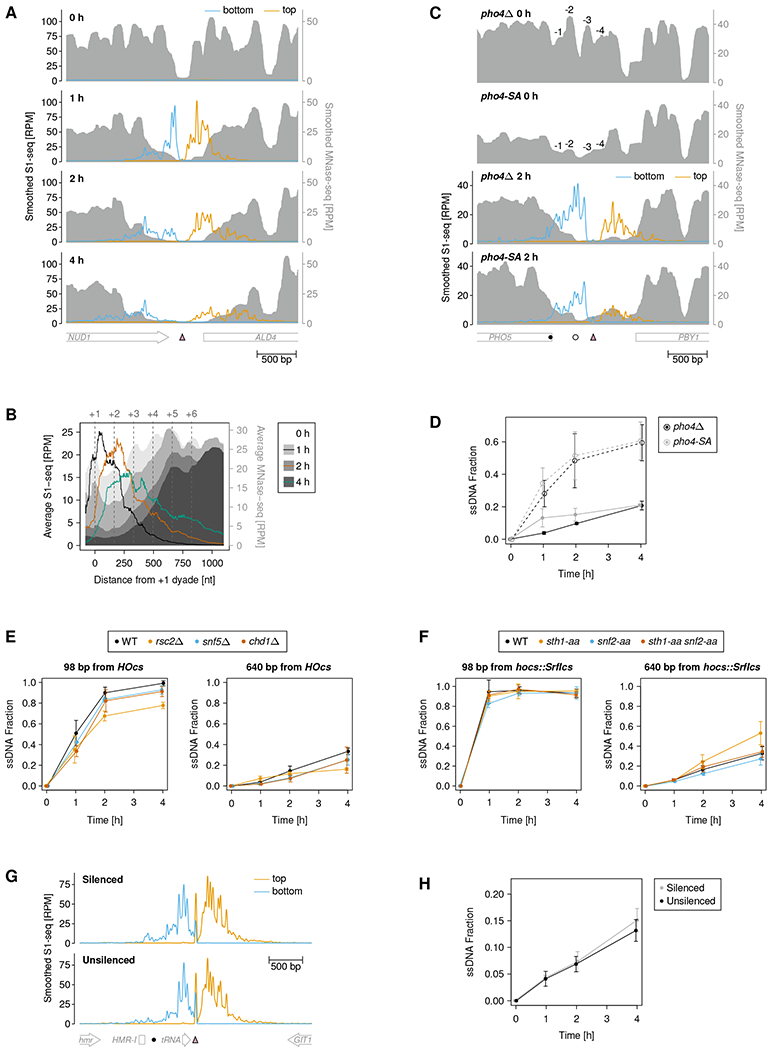
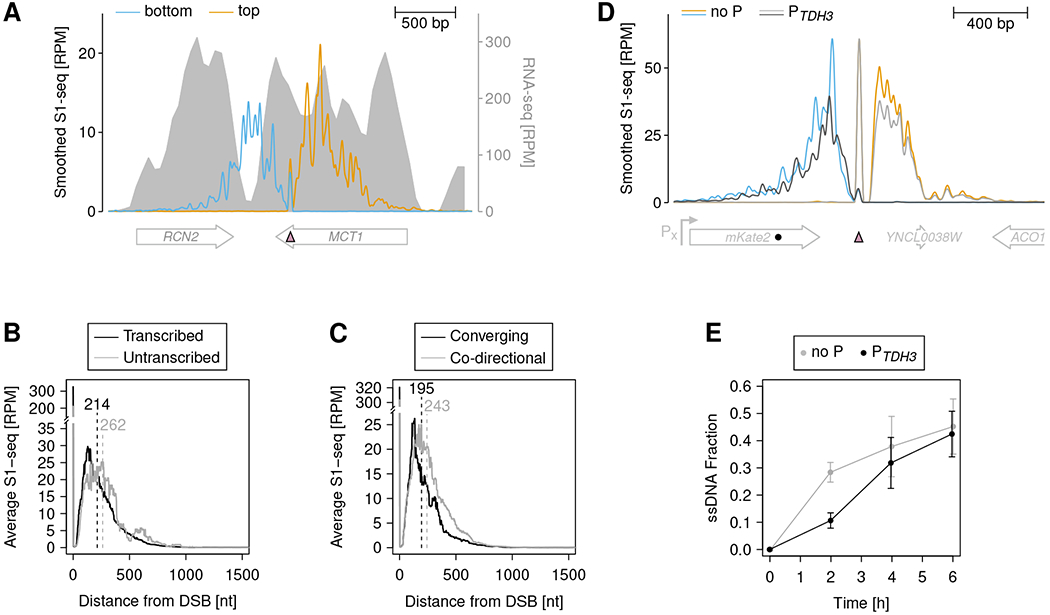
DNA double-strand breaks (DSBs) are cytotoxic genome lesions that must be accurately and efficiently repaired to ensure genome integrity. In yeast, the Mre11-Rad50-Xrs2 (MRX) complex nicks 5'-terminated DSB ends to initiate nucleolytic processing of DSBs for repair by homologous recombination. How MRX-DNA interactions support 5' strand-specific nicking and how nicking is influenced by the chromatin context have remained elusive. Using a deep sequencing-based assay, we mapped MRX nicks at single-nucleotide resolution next to multiple DSBs in the yeast genome. We observed that the DNA end-binding Ku70-Ku80 complex directed DSB-proximal nicks and that repetitive MRX cleavage extended the length of resection tracts. We identified a sequence motif and a DNA meltability profile that is preferentially nicked by MRX. Furthermore, we found that nucleosomes as well as transcription impeded MRX incisions. Our findings suggest that local DNA sequence and chromatin features shape the activity of this central DSB repair complex.
Keywords: CtIP; DNA double-strand break; DNA repair; Mre11; Sae2; chromatin; homologous recombination; resection.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
