

Preclinical proof of concept for VivoVec, a lentiviral-based platform for in vivo CAR T-cell engineering
- PMID: 36918221
- PMCID: PMC10016276
- DOI: 10.1136/jitc-2022-006292
Preclinical proof of concept for VivoVec, a lentiviral-based platform for in vivo CAR T-cell engineering
Abstract
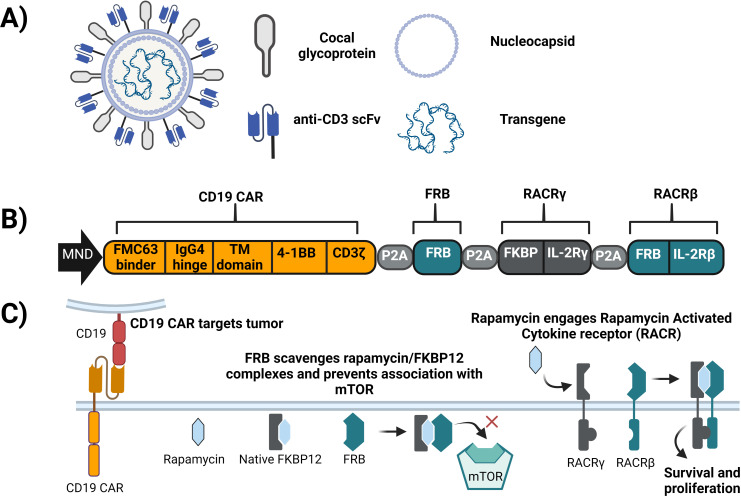
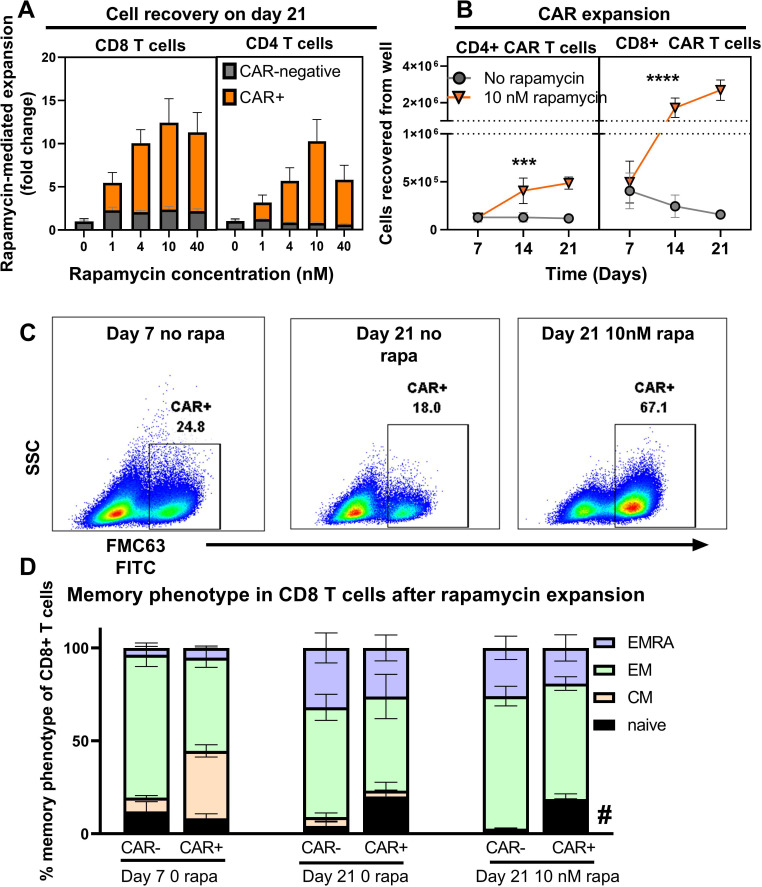
Background: Chimeric antigen receptor (CAR) T-cell therapies have demonstrated transformational outcomes in the treatment of B-cell malignancies, but their widespread use is hindered by technical and logistical challenges associated with ex vivo cell manufacturing. To overcome these challenges, we developed VivoVec, a lentiviral vector-based platform for in vivo engineering of T cells. UB-VV100, a VivoVec clinical candidate for the treatment of B-cell malignancies, displays an anti-CD3 single-chain variable fragment (scFv) on the surface and delivers a genetic payload that encodes a second-generation CD19-targeted CAR along with a rapamycin-activated cytokine receptor (RACR) system designed to overcome the need for lymphodepleting chemotherapy in supporting successful CAR T-cell expansion and persistence. In the presence of exogenous rapamycin, non-transduced immune cells are suppressed, while the RACR system in transduced cells converts rapamycin binding to an interleukin (IL)-2/IL-15 signal to promote proliferation.
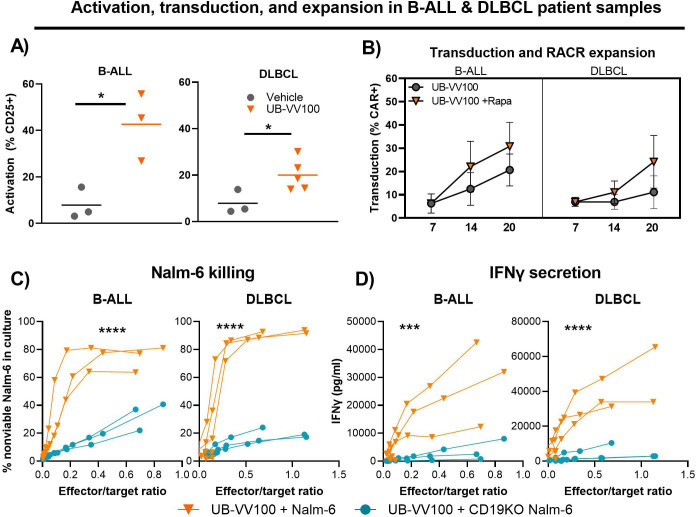
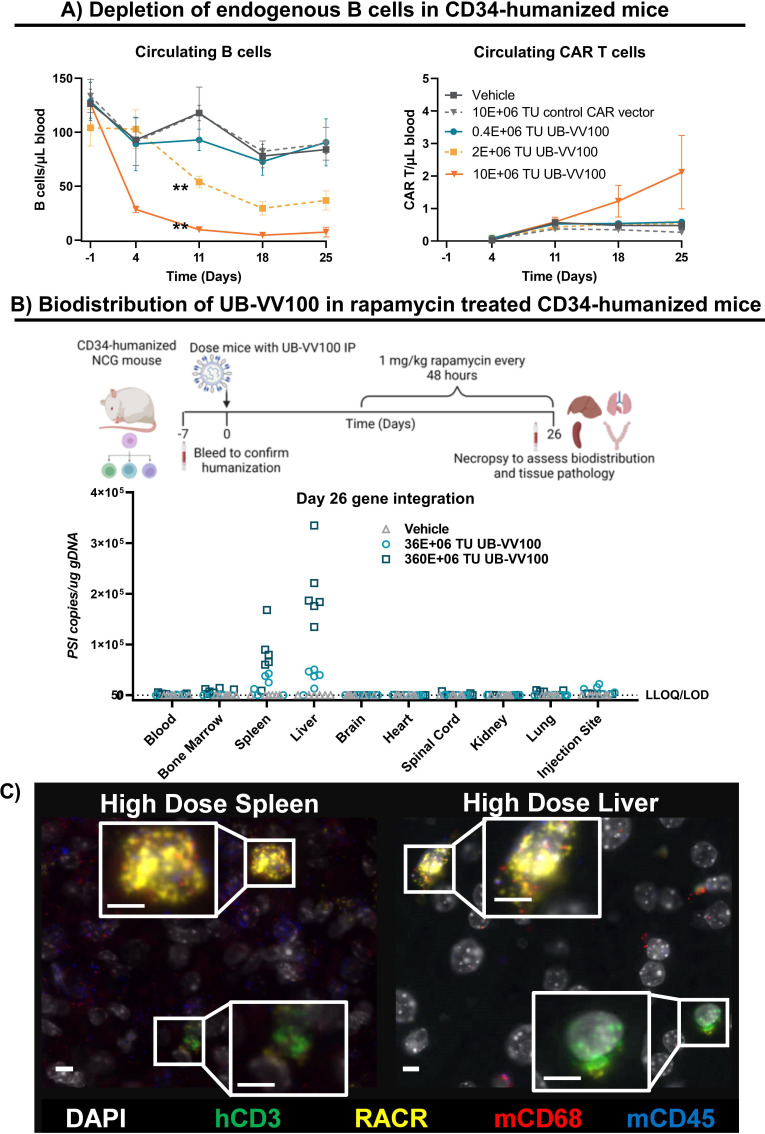
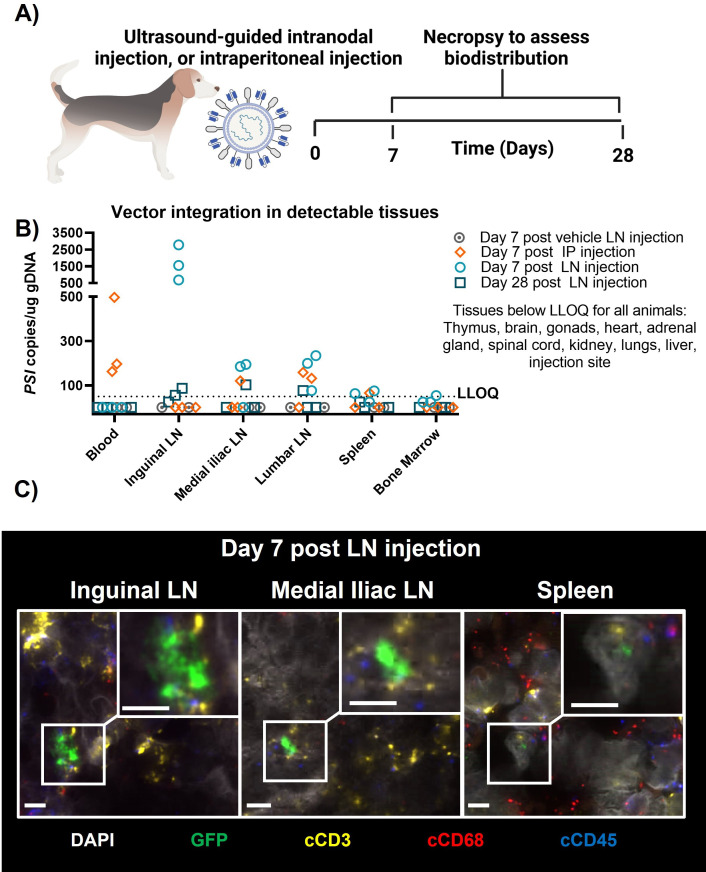
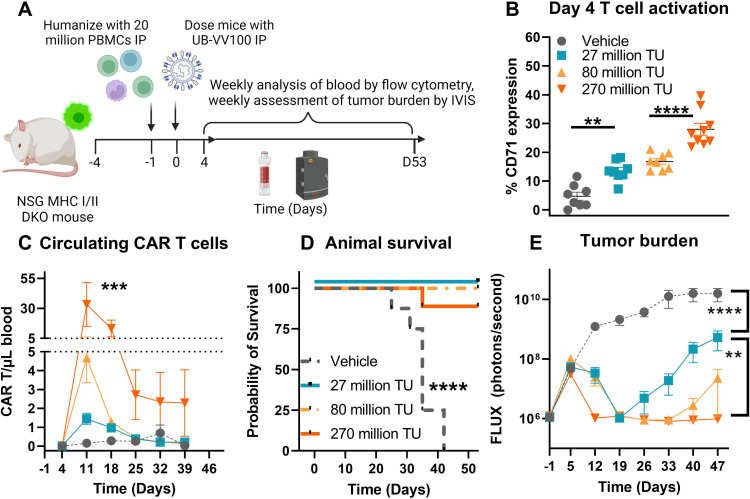
Methods: UB-VV100 was administered to peripheral blood mononuclear cells (PBMCs) from healthy donors and from patients with B-cell malignancy without additional stimulation. Cultures were assessed for CAR T-cell transduction and function. Biodistribution was evaluated in CD34-humanized mice and in canines. In vivo efficacy was evaluated against normal B cells in CD34-humanized mice and against systemic tumor xenografts in PBMC-humanized mice.
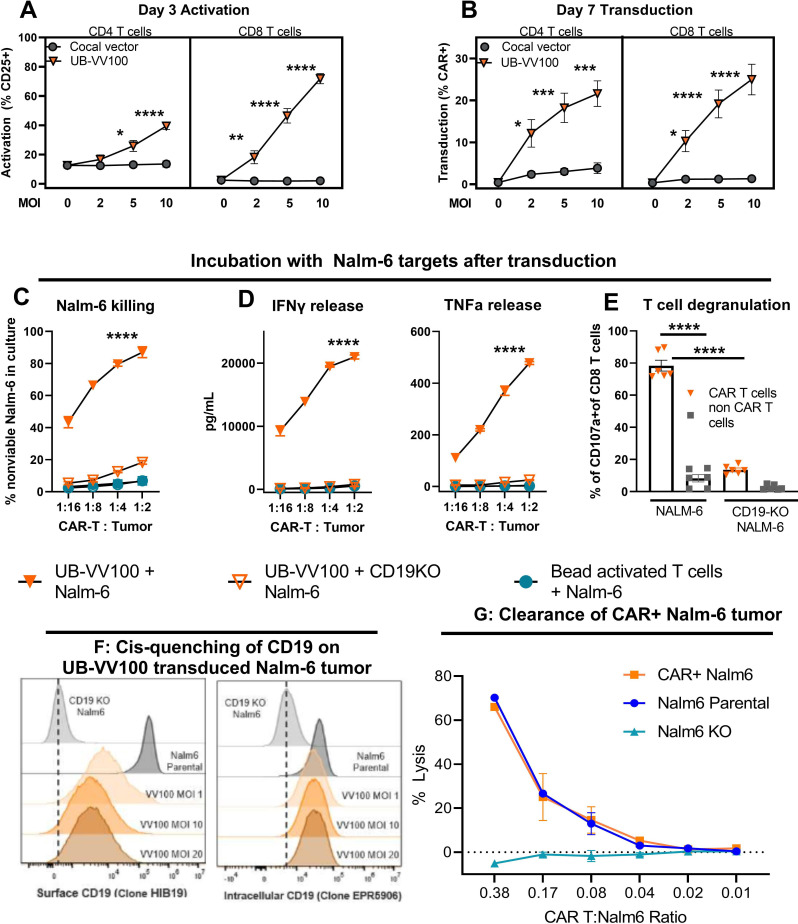
Results: In vitro, administration of UB-VV100 resulted in dose-dependent and anti-CD3 scFv-dependent T-cell activation and CAR T-cell transduction. The resulting CAR T cells exhibited selective expansion in rapamycin and antigen-dependent activity against malignant B-cell targets. In humanized mouse and canine studies, UB-VV100 demonstrated a favorable biodistribution profile, with transduction events limited to the immune compartment after intranodal or intraperitoneal administration. Administration of UB-VV100 to humanized mice engrafted with B-cell tumors resulted in CAR T-cell transduction, expansion, and elimination of systemic malignancy.
Conclusions: These findings demonstrate that UB-VV100 generates functional CAR T cells in vivo, which could expand patient access to CAR T technology in both hematological and solid tumors without the need for ex vivo cell manufacturing.
Keywords: Cell Engineering; Drug Evaluation, Preclinical; Immunotherapy; Receptors, Chimeric Antigen; Translational Medical Research.
© Author(s) (or their employer(s)) 2023. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: All authors hold equity with Umoja Biopharma.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources