

The roles of bone remodeling in normal hematopoiesis and age-related hematological malignancies
- PMID: 36918531
- PMCID: PMC10014945
- DOI: 10.1038/s41413-023-00249-w
The roles of bone remodeling in normal hematopoiesis and age-related hematological malignancies
Abstract
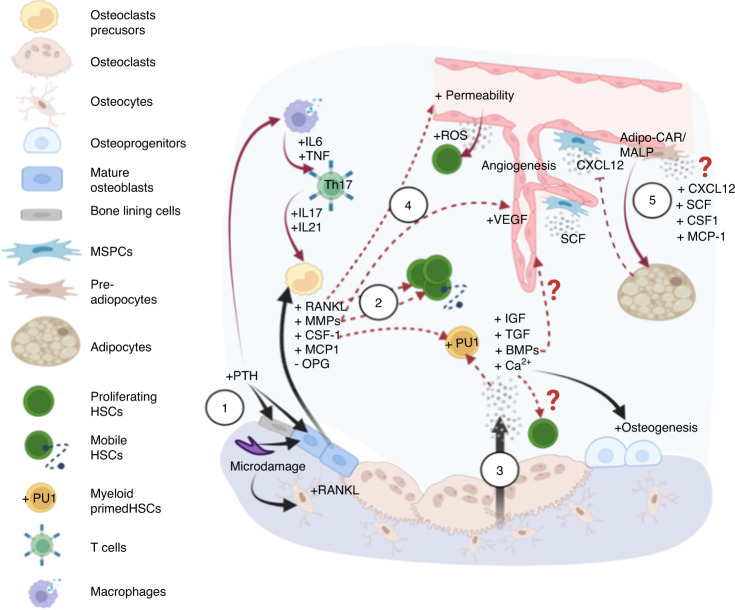
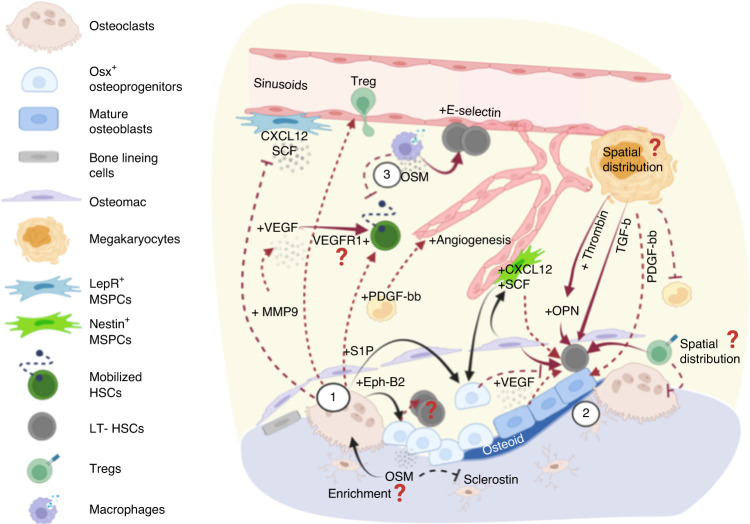
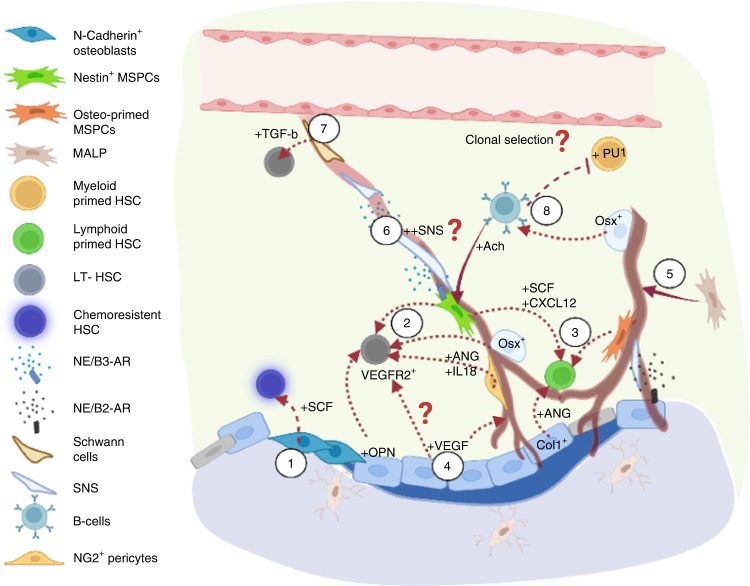
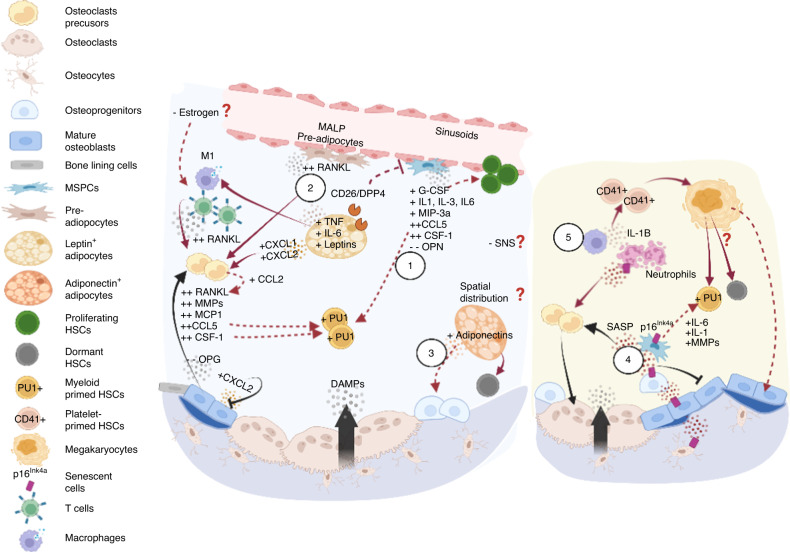
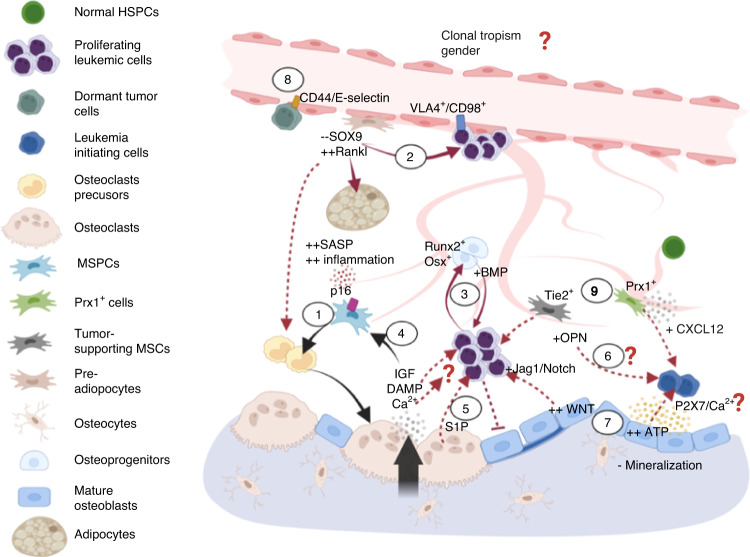
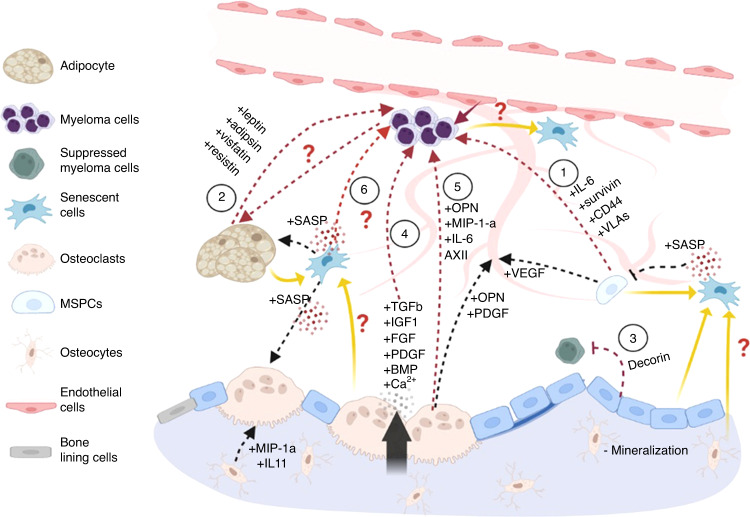
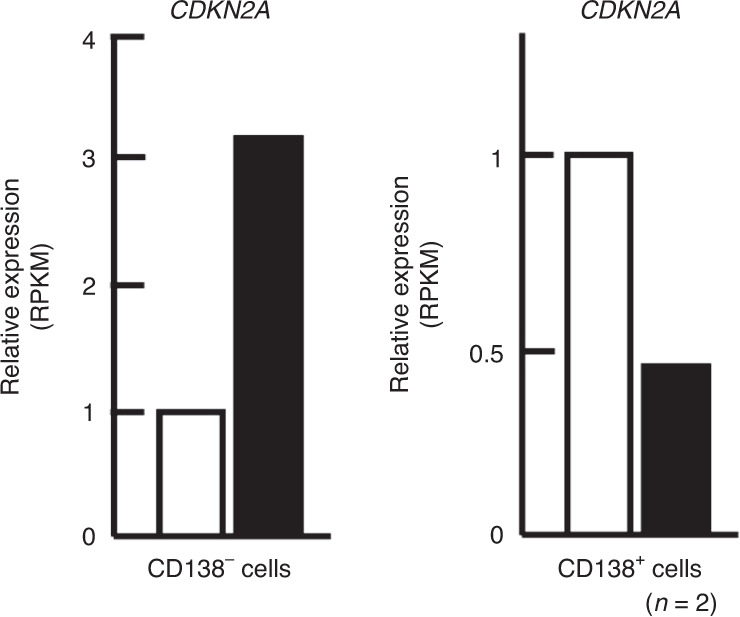
Prior research establishing that bone interacts in coordination with the bone marrow microenvironment (BMME) to regulate hematopoietic homeostasis was largely based on analyses of individual bone-associated cell populations. Recent advances in intravital imaging has suggested that the expansion of hematopoietic stem cells (HSCs) and acute myeloid leukemia cells is restricted to bone marrow microdomains during a distinct stage of bone remodeling. These findings indicate that dynamic bone remodeling likely imposes additional heterogeneity within the BMME to yield differential clonal responses. A holistic understanding of the role of bone remodeling in regulating the stem cell niche and how these interactions are altered in age-related hematological malignancies will be critical to the development of novel interventions. To advance this understanding, herein, we provide a synopsis of the cellular and molecular constituents that participate in bone turnover and their known connections to the hematopoietic compartment. Specifically, we elaborate on the coupling between bone remodeling and the BMME in homeostasis and age-related hematological malignancies and after treatment with bone-targeting approaches. We then discuss unresolved questions and ambiguities that remain in the field.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
