

Metabolic landscape in cardiac aging: insights into molecular biology and therapeutic implications
- PMID: 36918543
- PMCID: PMC10015017
- DOI: 10.1038/s41392-023-01378-8
Metabolic landscape in cardiac aging: insights into molecular biology and therapeutic implications
Abstract
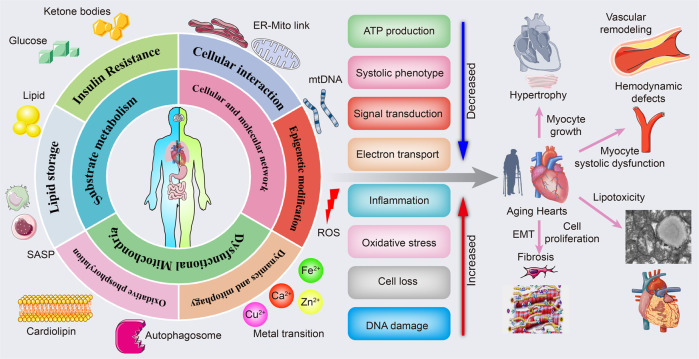
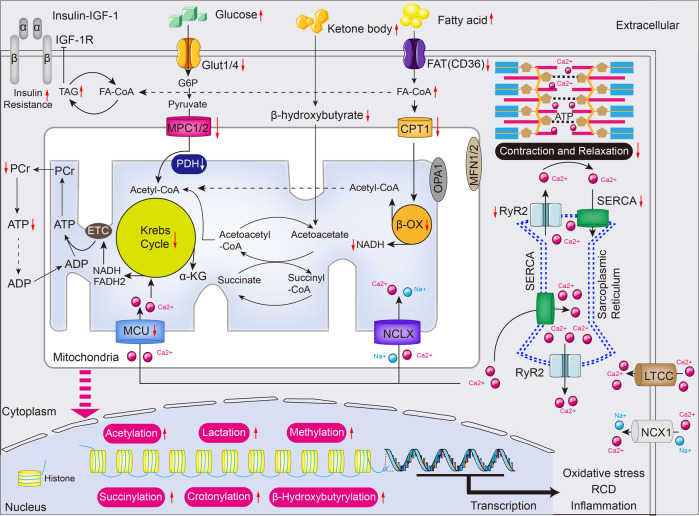
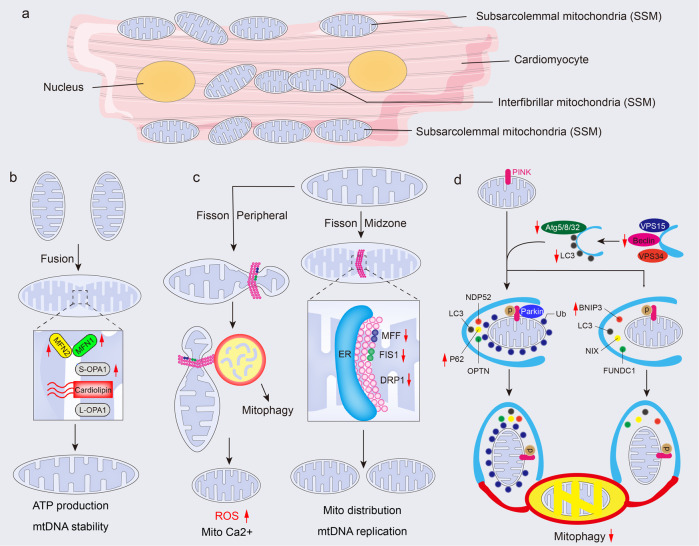
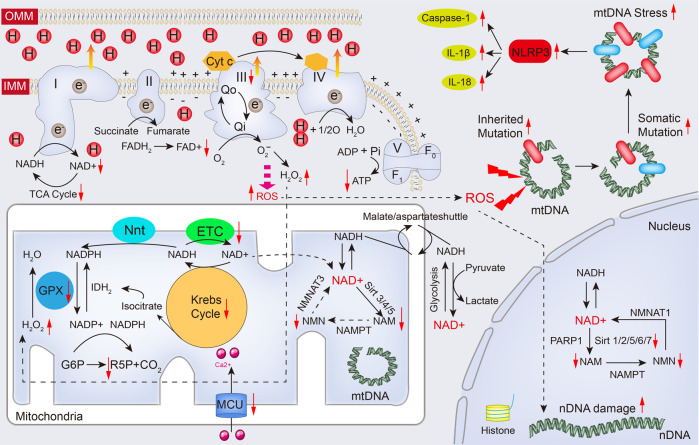
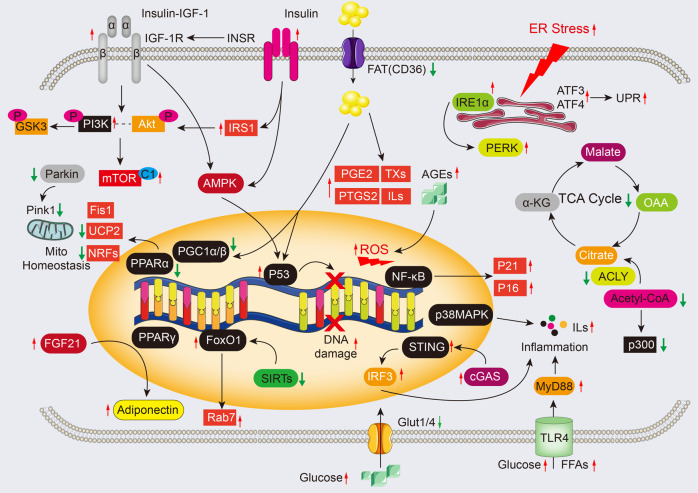
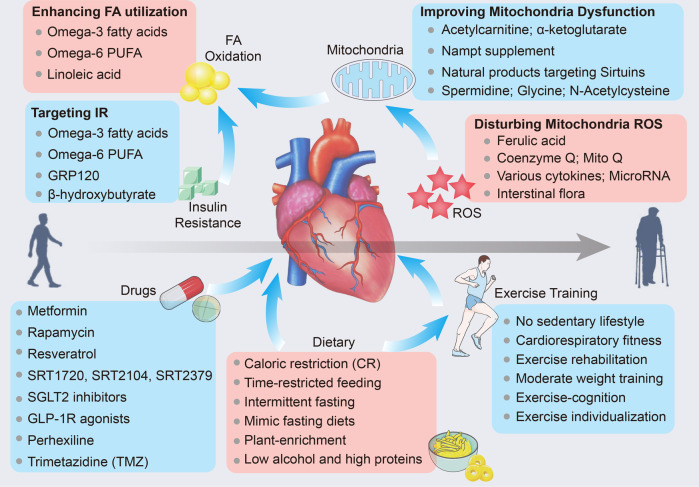
Cardiac aging is evident by a reduction in function which subsequently contributes to heart failure. The metabolic microenvironment has been identified as a hallmark of malignancy, but recent studies have shed light on its role in cardiovascular diseases (CVDs). Various metabolic pathways in cardiomyocytes and noncardiomyocytes determine cellular senescence in the aging heart. Metabolic alteration is a common process throughout cardiac degeneration. Importantly, the involvement of cellular senescence in cardiac injuries, including heart failure and myocardial ischemia and infarction, has been reported. However, metabolic complexity among human aging hearts hinders the development of strategies that targets metabolic susceptibility. Advances over the past decade have linked cellular senescence and function with their metabolic reprogramming pathway in cardiac aging, including autophagy, oxidative stress, epigenetic modifications, chronic inflammation, and myocyte systolic phenotype regulation. In addition, metabolic status is involved in crucial aspects of myocardial biology, from fibrosis to hypertrophy and chronic inflammation. However, further elucidation of the metabolism involvement in cardiac degeneration is still needed. Thus, deciphering the mechanisms underlying how metabolic reprogramming impacts cardiac aging is thought to contribute to the novel interventions to protect or even restore cardiac function in aging hearts. Here, we summarize emerging concepts about metabolic landscapes of cardiac aging, with specific focuses on why metabolic profile alters during cardiac degeneration and how we could utilize the current knowledge to improve the management of cardiac aging.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
