

SARS-CoV-2 receptor-binding domain deep mutational AlphaFold2 structures
- PMID: 36918581
- PMCID: PMC10013278
- DOI: 10.1038/s41597-023-02035-z
SARS-CoV-2 receptor-binding domain deep mutational AlphaFold2 structures
Abstract
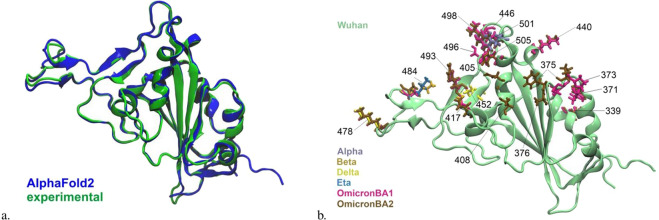
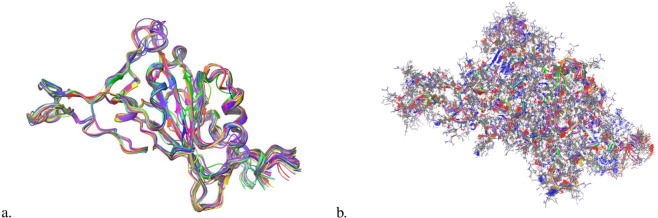
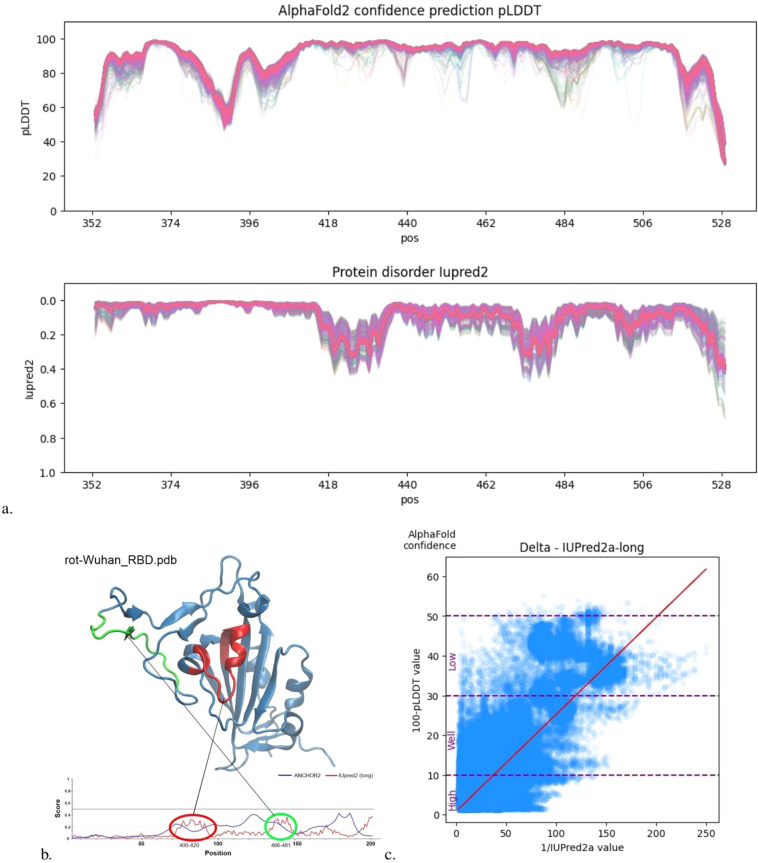
Leveraging recent advances in computational modeling of proteins with AlphaFold2 (AF2) we provide a complete curated data set of all single mutations from each of the 7 main SARS-CoV-2 lineages spike protein receptor binding domain (RBD) resulting in 3819X7 = 26733 PDB structures. We visualize the generated structures and show that AF2 pLDDT values are correlated with state-of-the-art disorder approximations, implying some internal protein dynamics are also captured by the model. Joint increasing mutational coverage of both structural and phenotype data coupled with advances in machine learning can be leveraged to accelerate virology research, specifically future variant prediction. We hope this data release can offer assistance into further understanding of the local and global mutational landscape of SARS-CoV-2 as well as provide insight into the biological understanding that 3D structure acts as a bridge between protein genotype and phenotype.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
