

Synthetic enforcement of STING signaling in cancer cells appropriates the immune microenvironment for checkpoint inhibitor therapy
- PMID: 36921054
- PMCID: PMC10017047
- DOI: 10.1126/sciadv.add8564
Synthetic enforcement of STING signaling in cancer cells appropriates the immune microenvironment for checkpoint inhibitor therapy
Abstract
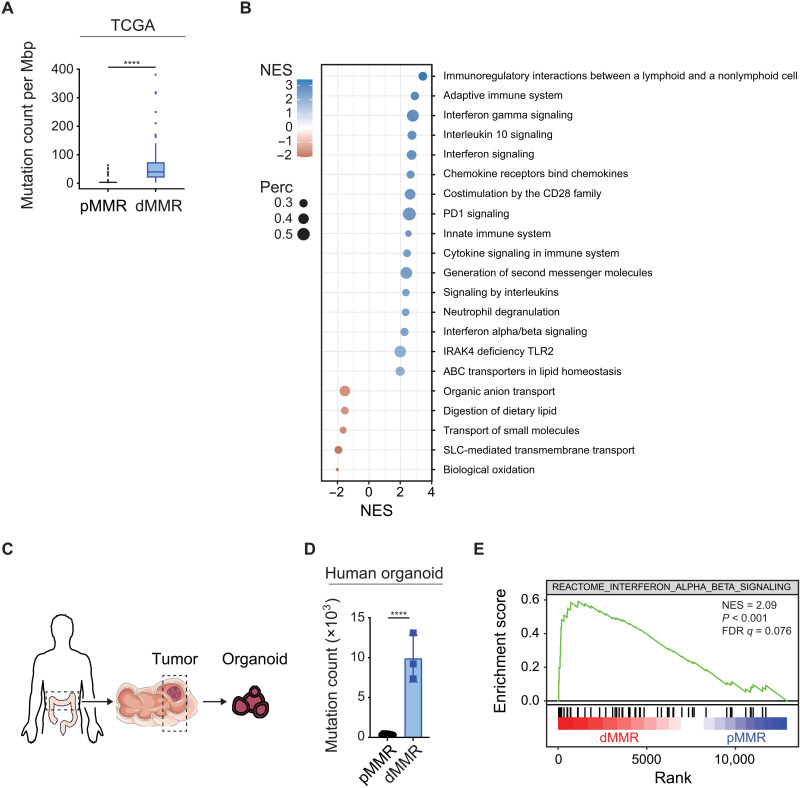
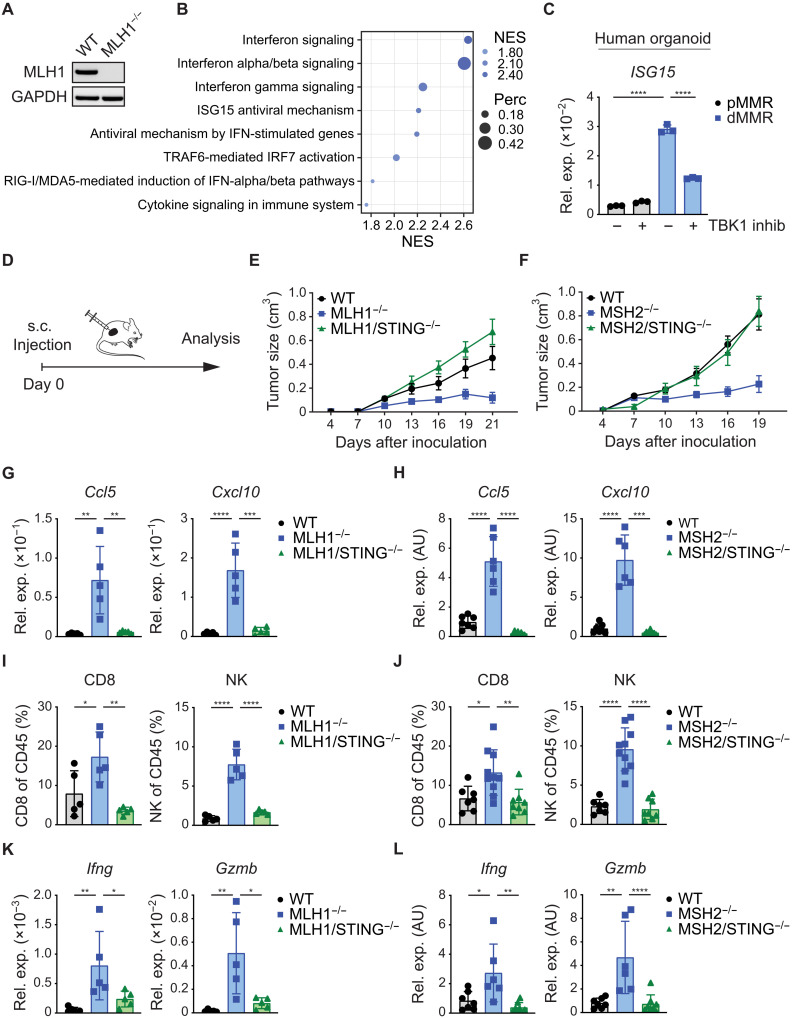
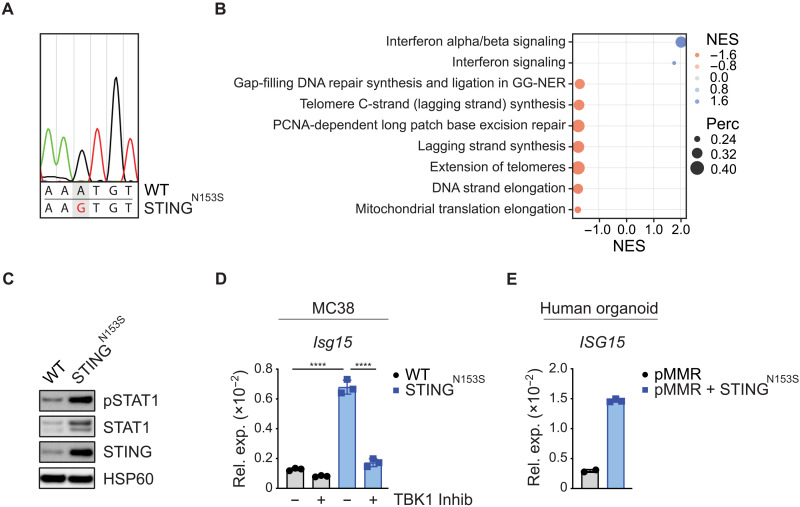
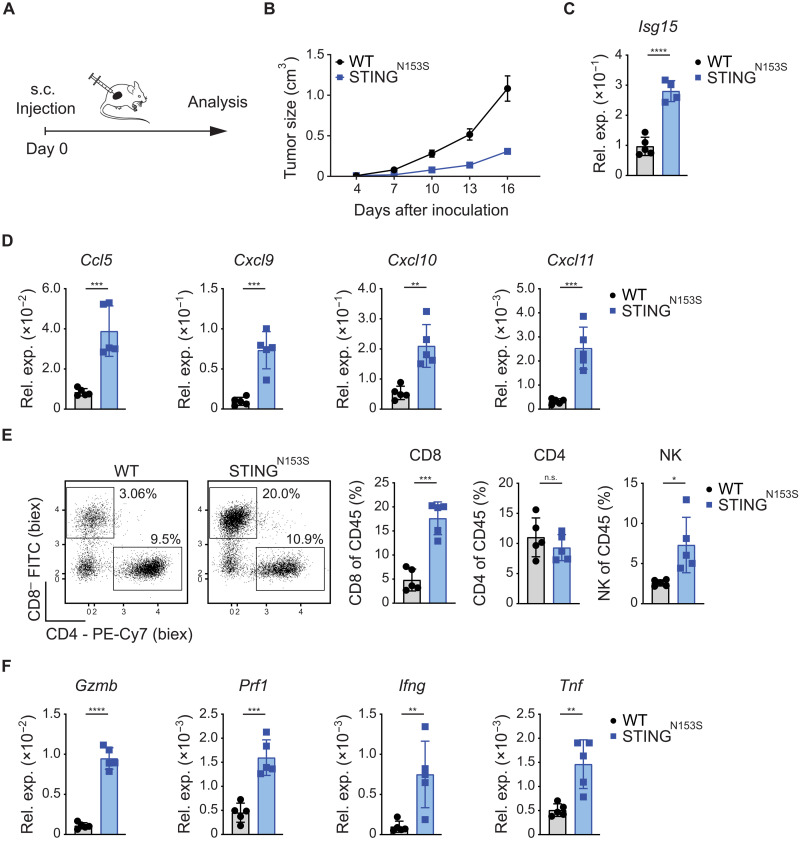
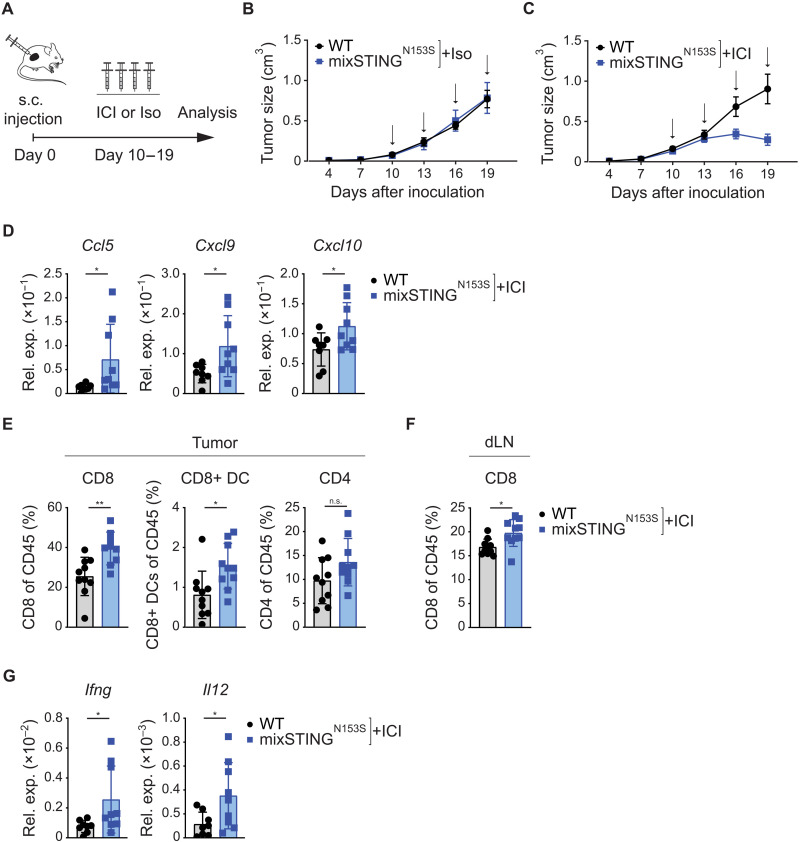
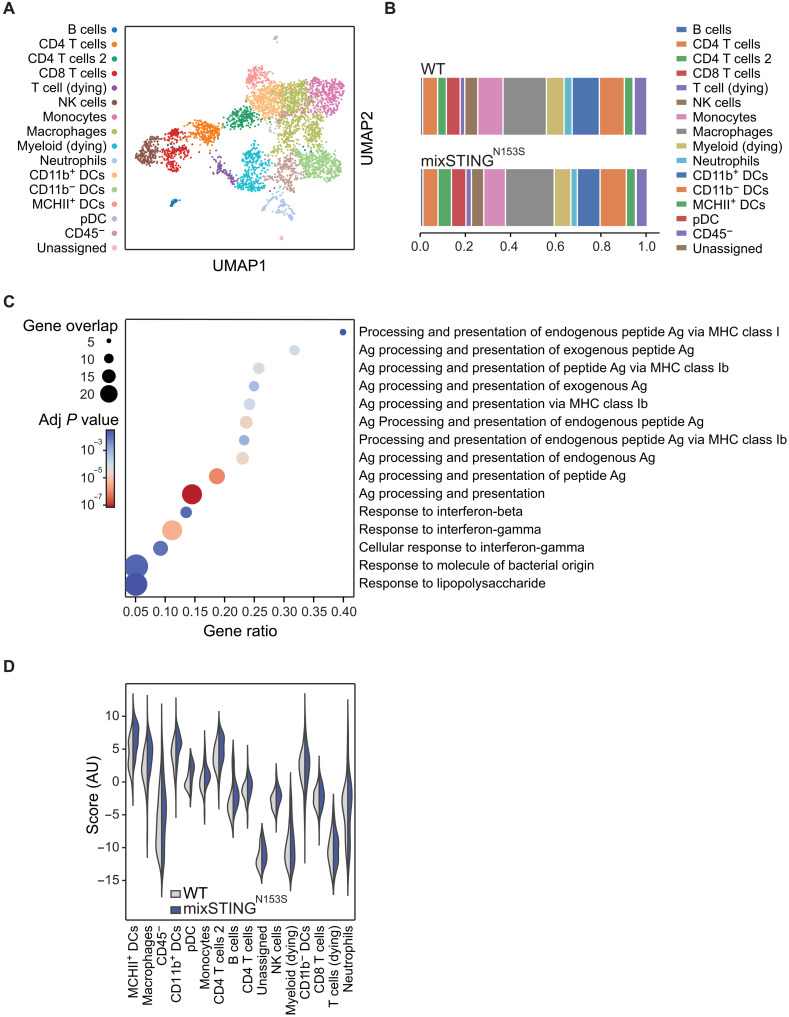
Immune checkpoint inhibitors (ICIs) enhance anticancer immunity by releasing repressive signals into tumor microenvironments (TMEs). To be effective, ICIs require preexisting immunologically "hot" niches for tumor antigen presentation and lymphocyte recruitment. How the mutational landscape of cancer cells shapes these immunological niches remains poorly defined. We found in human and murine colorectal cancer (CRC) models that the superior antitumor immune response of mismatch repair (MMR)-deficient CRC required tumor cell-intrinsic activation of cGAS-STING signaling triggered by genomic instability. Subsequently, we synthetically enforced STING signaling in CRC cells with intact MMR signaling using constitutively active STING variants. Even in MMR-proficient CRC, genetically encoded gain-of-function STING was sufficient to induce cancer cell-intrinsic interferon signaling, local activation of antigen-presenting cells, recruitment of effector lymphocytes, and sensitization of previously "cold" TMEs to ICI therapy in vivo. Thus, our results introduce a rational strategy for modulating cancer cell-intrinsic programs via engineered STING enforcement to sensitize resistant tumors to ICI responsiveness.
Figures
References
-
- D. S. Chen, I. Mellman, Elements of cancer immunity and the cancer-immune set point. Nature 541, 321–330 (2017). - PubMed
-
- J. Galon, D. Bruni, Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 18, 197–218 (2019). - PubMed
-
- D. T. Le, J. N. Durham, K. N. Smith, H. Wang, B. R. Bartlett, L. K. Aulakh, S. Lu, H. Kemberling, C. Wilt, B. S. Luber, F. Wong, N. S. Azad, A. A. Rucki, D. Laheru, R. Donehower, A. Zaheer, G. A. Fisher, T. S. Crocenzi, J. J. Lee, T. F. Greten, A. G. Duffy, K. K. Ciombor, A. D. Eyring, B. H. Lam, A. Joe, S. P. Kang, M. Holdhoff, L. Danilova, L. Cope, C. Meyer, S. Zhou, R. M. Goldberg, D. K. Armstrong, K. M. Bever, A. N. Fader, J. Taube, F. Housseau, D. Spetzler, N. Xiao, D. M. Pardoll, N. Papadopoulos, K. W. Kinzler, J. R. Eshleman, B. Vogelstein, R. A. Anders, L. A. Diaz Jr., Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017). - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
