

Genetic Modifiers of Cystic Fibrosis Lung Disease Severity: Whole-Genome Analysis of 7,840 Patients
- PMID: 36921087
- PMCID: PMC10595435
- DOI: 10.1164/rccm.202209-1653OC
Genetic Modifiers of Cystic Fibrosis Lung Disease Severity: Whole-Genome Analysis of 7,840 Patients
Abstract
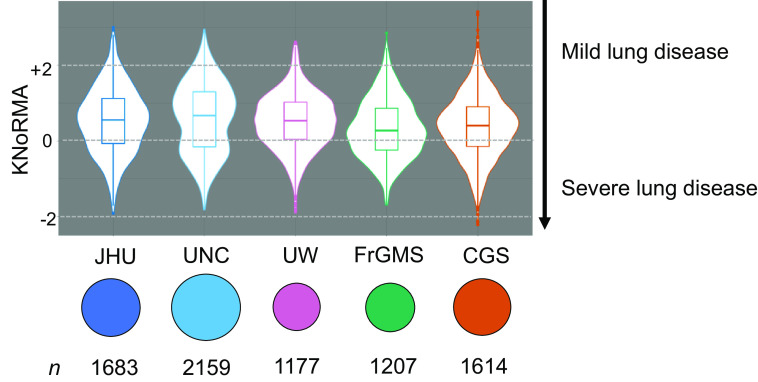
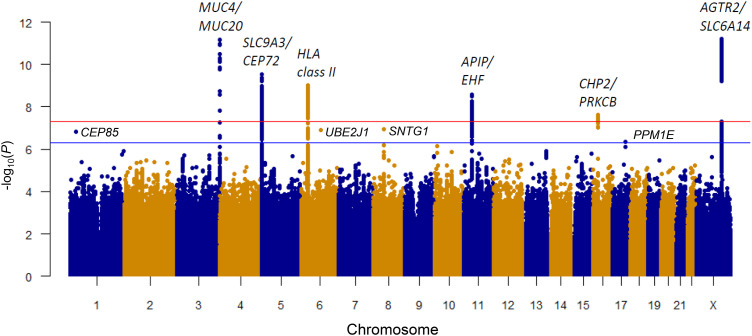
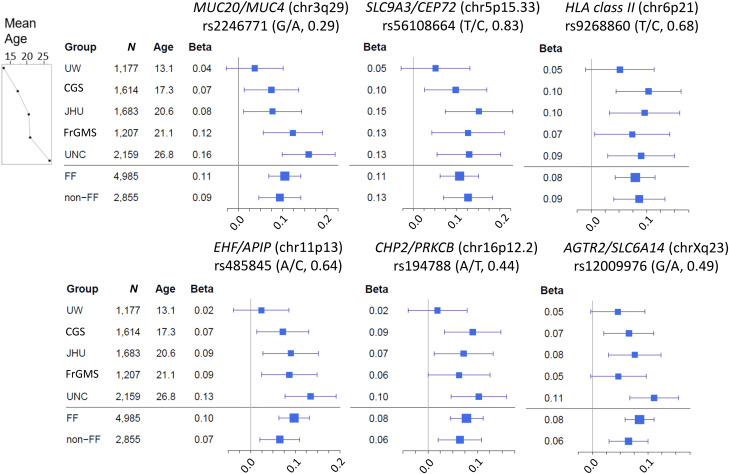
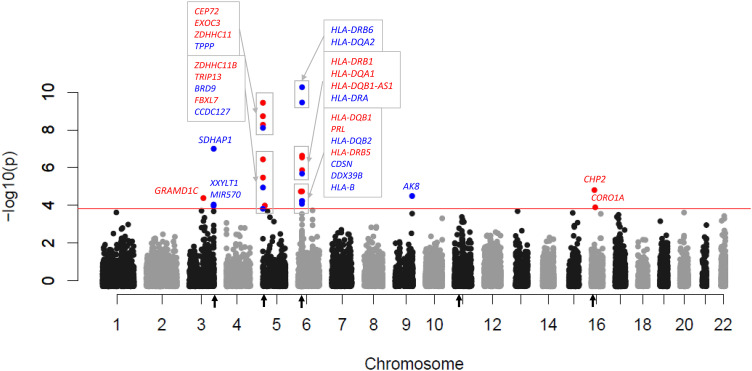
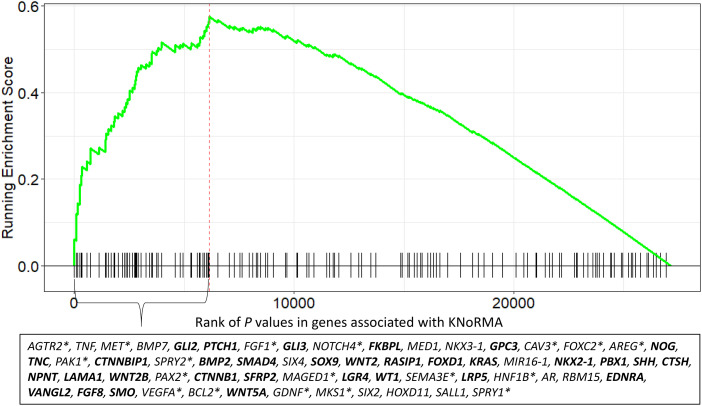
Rationale: Lung disease is the major cause of morbidity and mortality in persons with cystic fibrosis (pwCF). Variability in CF lung disease has substantial non-CFTR (CF transmembrane conductance regulator) genetic influence. Identification of genetic modifiers has prognostic and therapeutic importance. Objectives: Identify genetic modifier loci and genes/pathways associated with pulmonary disease severity. Methods: Whole-genome sequencing data on 4,248 unique pwCF with pancreatic insufficiency and lung function measures were combined with imputed genotypes from an additional 3,592 patients with pancreatic insufficiency from the United States, Canada, and France. This report describes association of approximately 15.9 million SNPs using the quantitative Kulich normal residual mortality-adjusted (KNoRMA) lung disease phenotype in 7,840 pwCF using premodulator lung function data. Measurements and Main Results: Testing included common and rare SNPs, transcriptome-wide association, gene-level, and pathway analyses. Pathway analyses identified novel associations with genes that have key roles in organ development, and we hypothesize that these genes may relate to dysanapsis and/or variability in lung repair. Results confirmed and extended previous genome-wide association study findings. These whole-genome sequencing data provide finely mapped genetic information to support mechanistic studies. No novel primary associations with common single variants or rare variants were found. Multilocus effects at chr5p13 (SLC9A3/CEP72) and chr11p13 (EHF/APIP) were identified. Variant effect size estimates at associated loci were consistently ordered across the cohorts, indicating possible age or birth cohort effects. Conclusions: This premodulator genomic, transcriptomic, and pathway association study of 7,840 pwCF will facilitate mechanistic and postmodulator genetic studies and the development of novel therapeutics for CF lung disease.
Keywords: GWAS/TWAS; cystic fibrosis; lung disease severity; pathway analyses; whole-genome sequencing.
Figures
Comment in
-
Who Modifies the Modifiers: A High-Resolution View of the Genetic Modifiers of Cystic Fibrosis.Am J Respir Crit Care Med. 2023 May 15;207(10):1261-1262. doi: 10.1164/rccm.202303-0468ED. Am J Respir Crit Care Med. 2023. PMID: 36961916 Free PMC article. No abstract available.
References
-
- O’Neal WK, Knowles MR. Cystic fibrosis disease modifiers: complex genetics defines the phenotypic diversity in a monogenic disease. Annu Rev Genomics Hum Genet . 2018;19:201–222. - PubMed
-
- Egan ME. Cystic fibrosis transmembrane conductance receptor modulator therapy in cystic fibrosis, an update. Curr Opin Pediatr . 2020;32:384–388. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
