

Spatial epigenome-transcriptome co-profiling of mammalian tissues
- PMID: 36922587
- PMCID: PMC10076218
- DOI: 10.1038/s41586-023-05795-1
Spatial epigenome-transcriptome co-profiling of mammalian tissues
Abstract
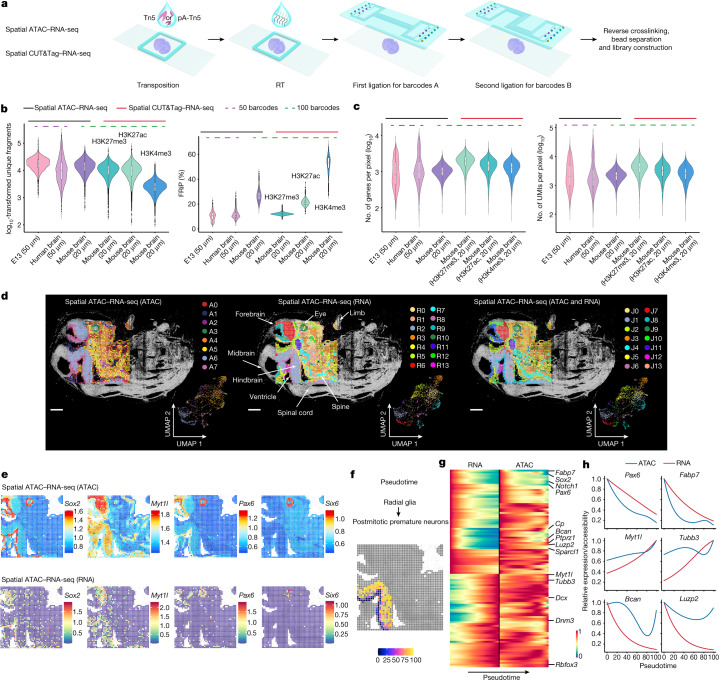
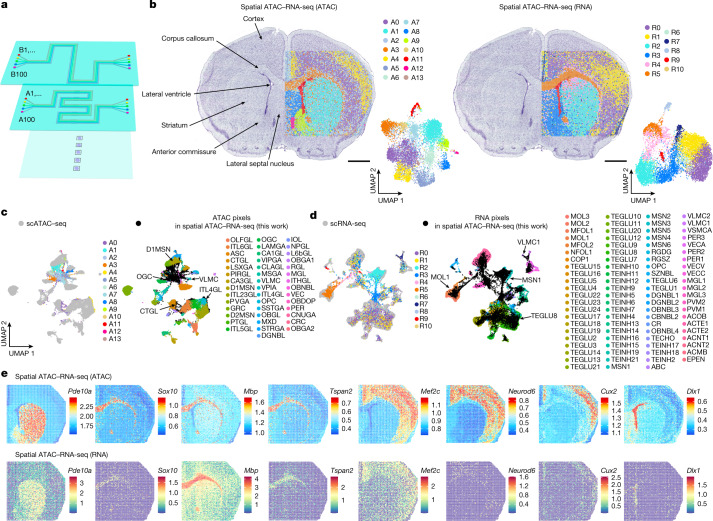
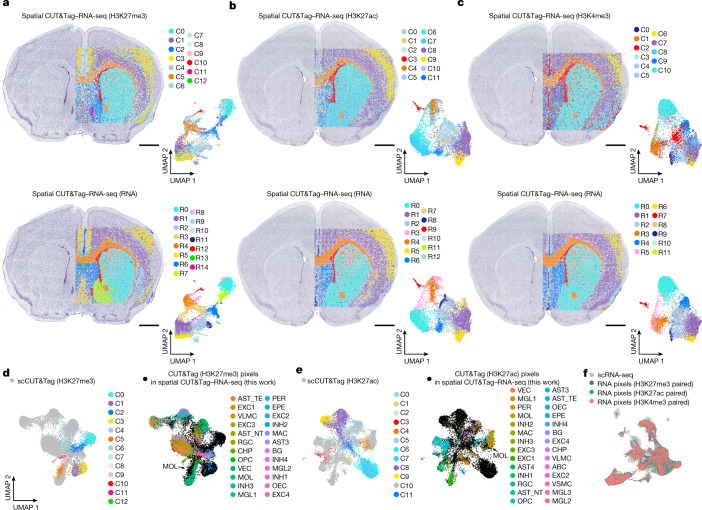
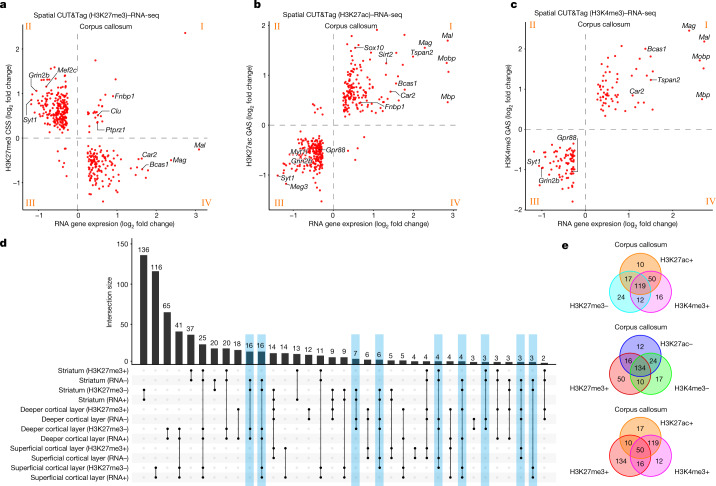
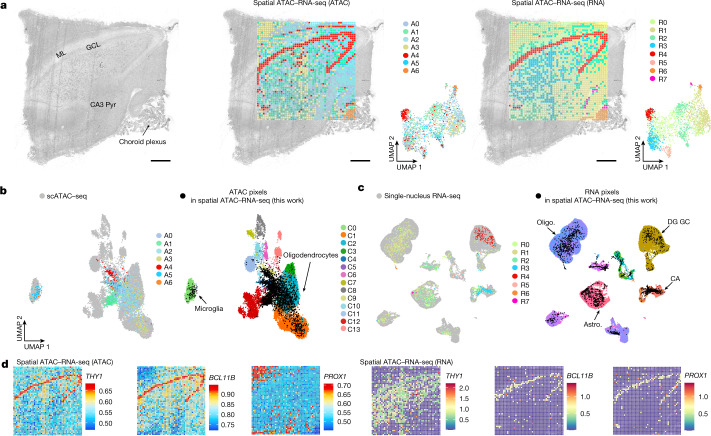
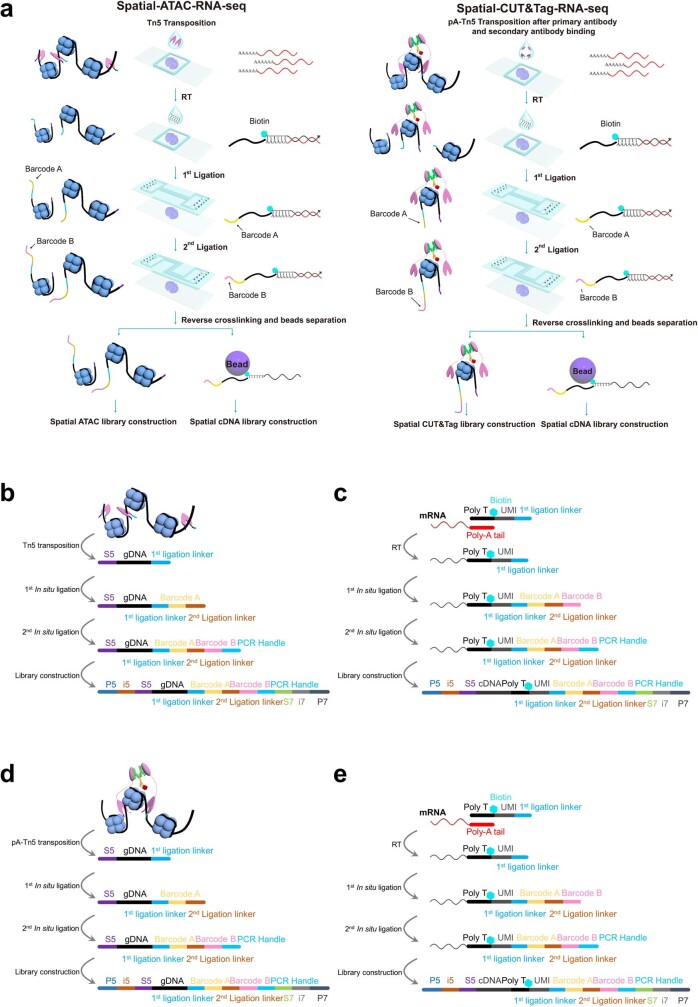
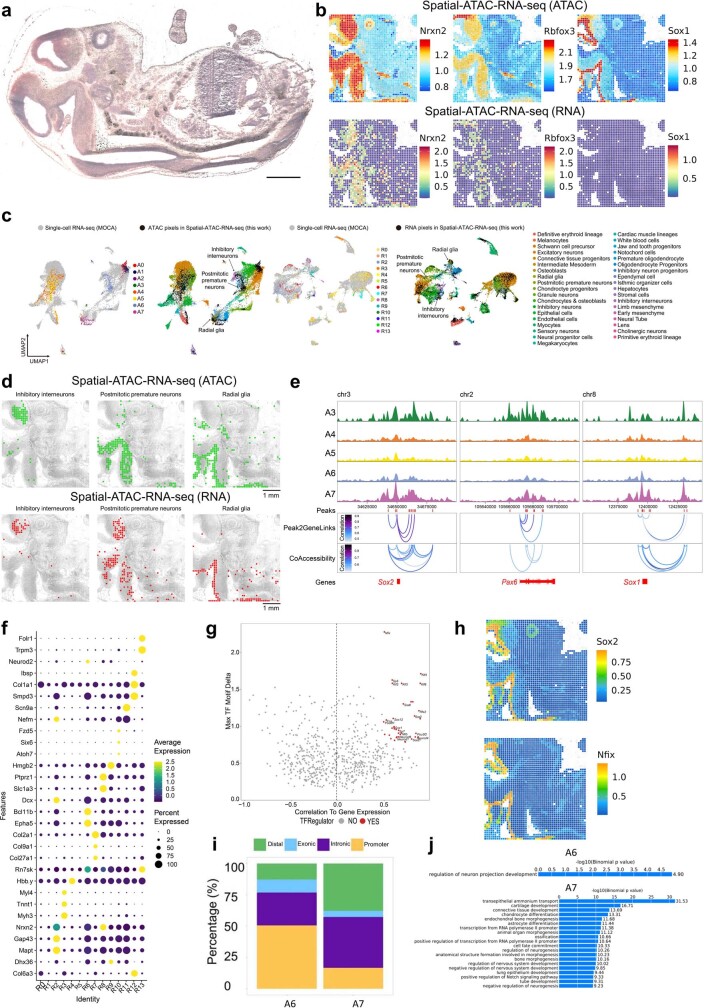
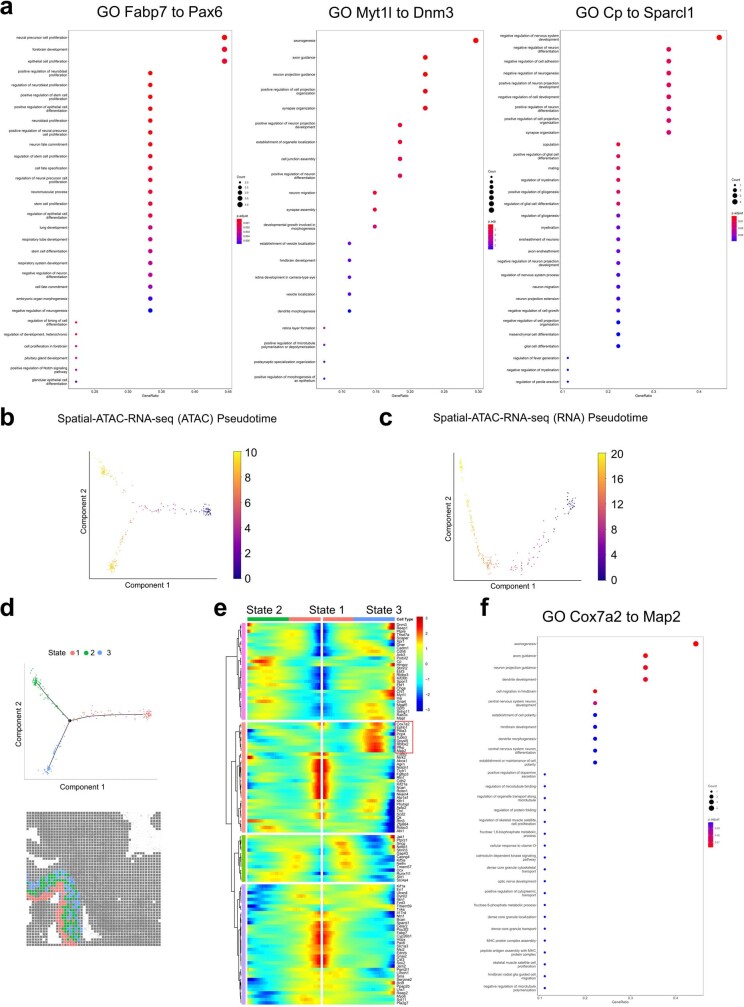
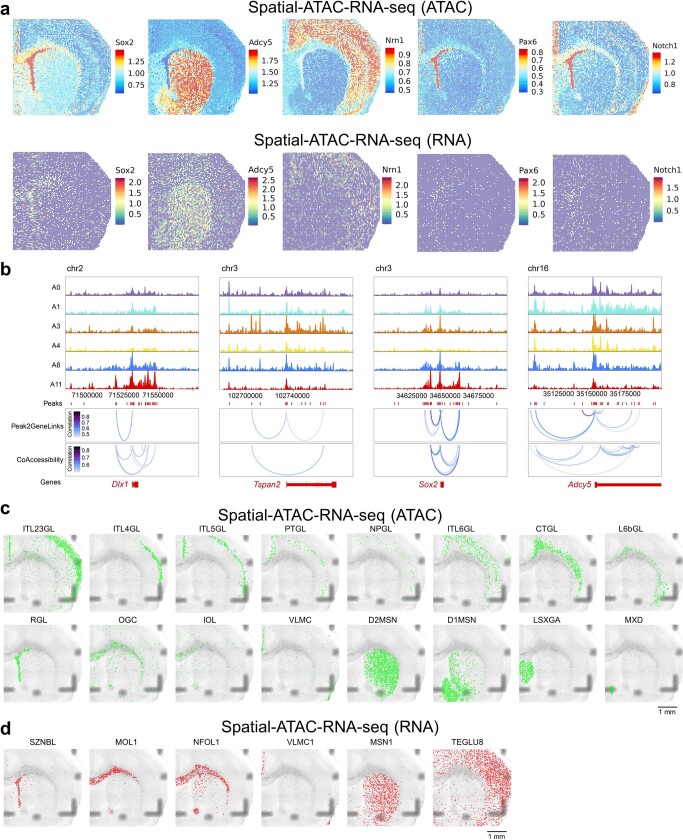
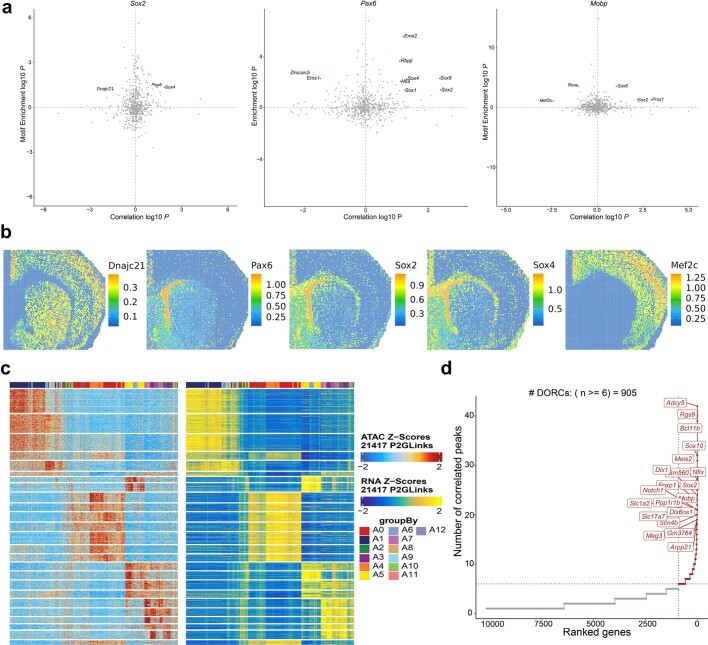
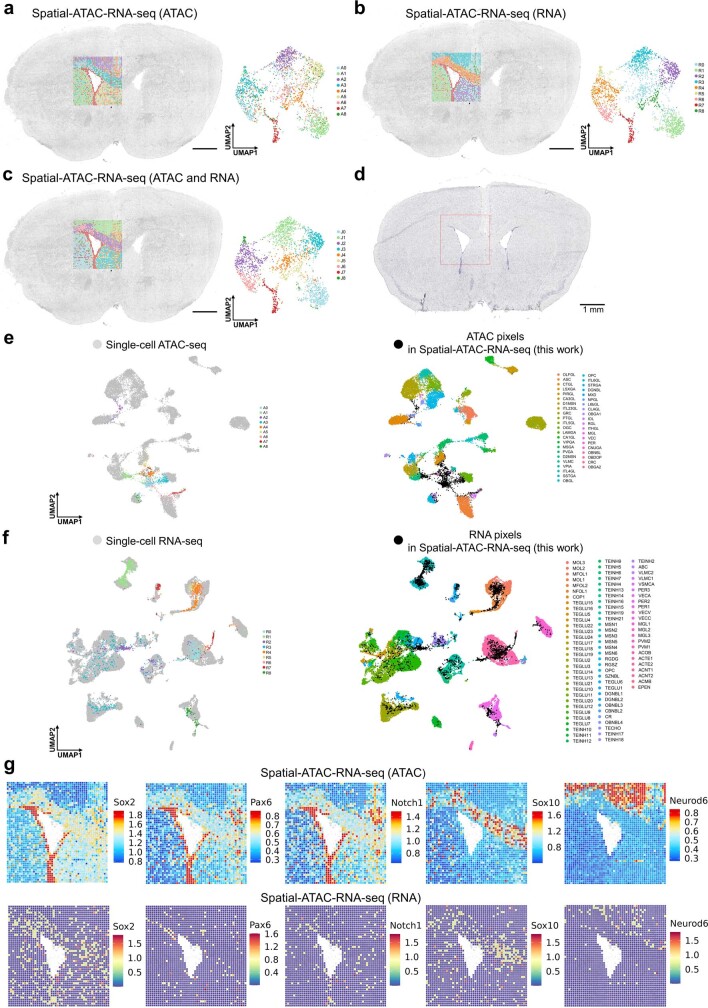
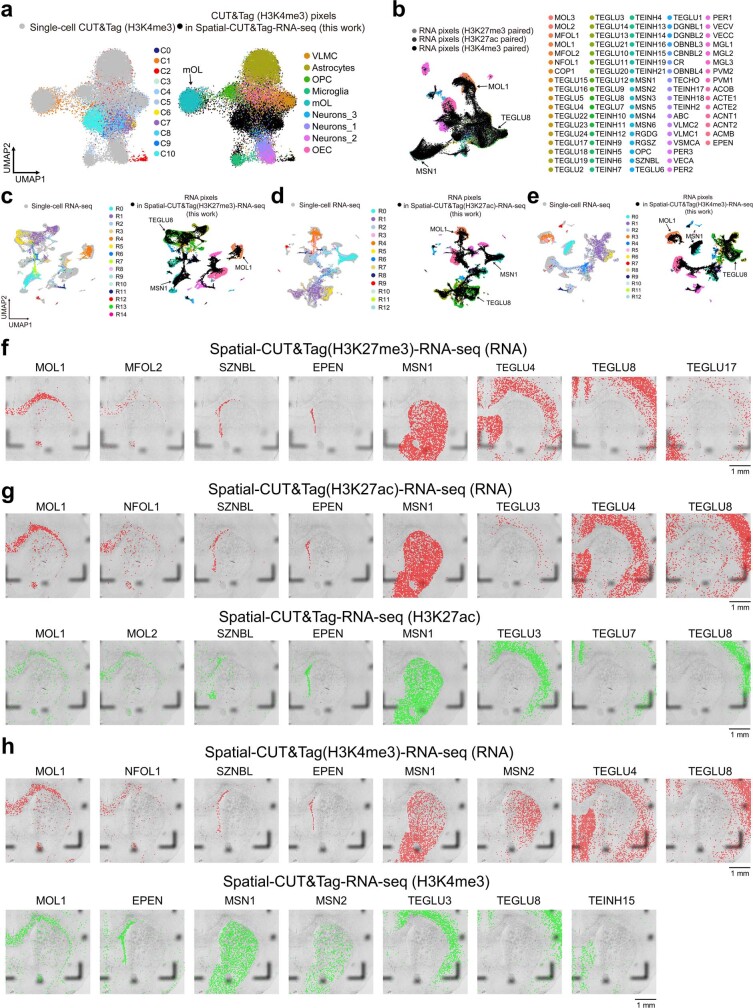
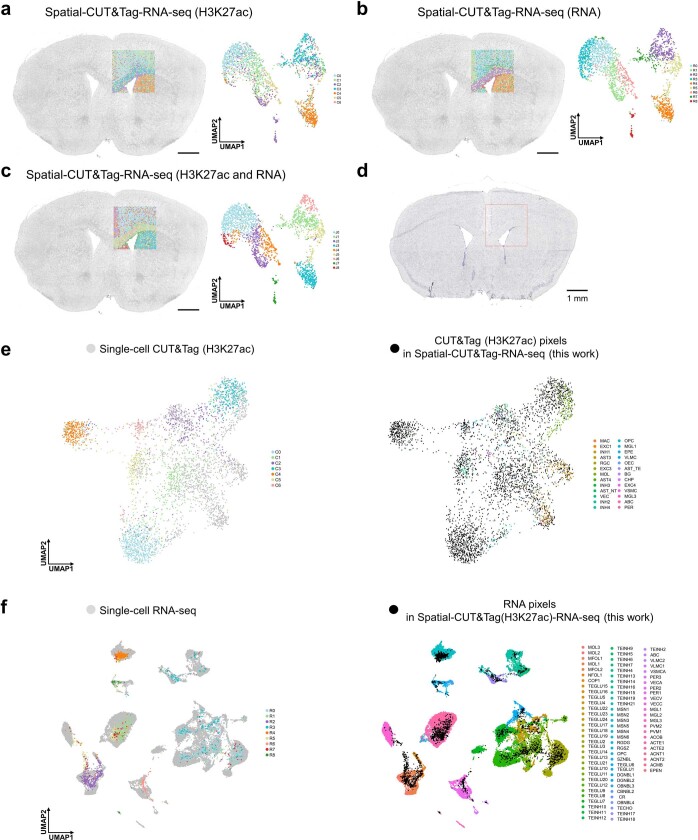
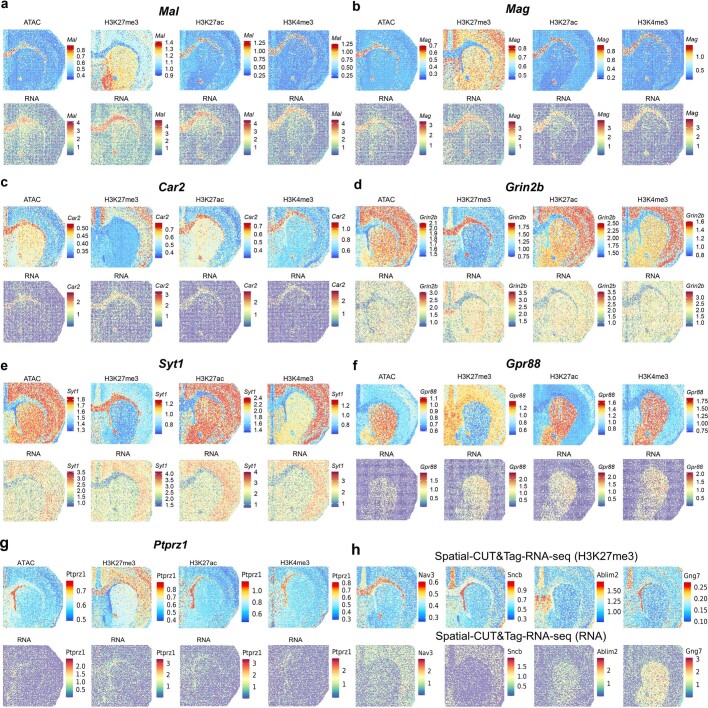
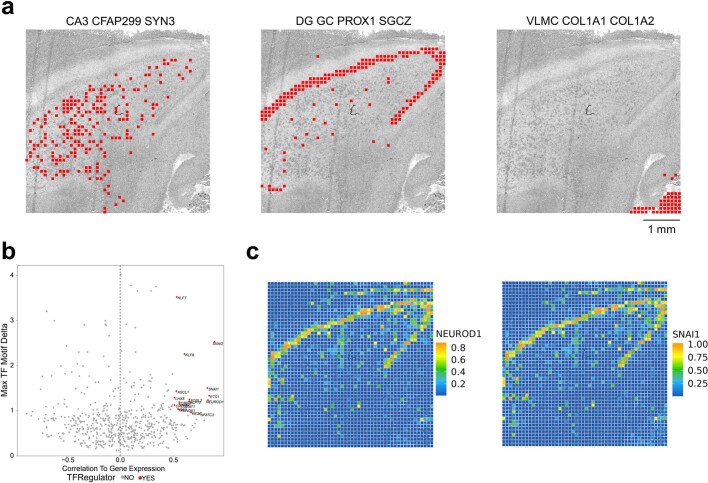
Emerging spatial technologies, including spatial transcriptomics and spatial epigenomics, are becoming powerful tools for profiling of cellular states in the tissue context1-5. However, current methods capture only one layer of omics information at a time, precluding the possibility of examining the mechanistic relationship across the central dogma of molecular biology. Here, we present two technologies for spatially resolved, genome-wide, joint profiling of the epigenome and transcriptome by cosequencing chromatin accessibility and gene expression, or histone modifications (H3K27me3, H3K27ac or H3K4me3) and gene expression on the same tissue section at near-single-cell resolution. These were applied to embryonic and juvenile mouse brain, as well as adult human brain, to map how epigenetic mechanisms control transcriptional phenotype and cell dynamics in tissue. Although highly concordant tissue features were identified by either spatial epigenome or spatial transcriptome we also observed distinct patterns, suggesting their differential roles in defining cell states. Linking epigenome to transcriptome pixel by pixel allows the uncovering of new insights in spatial epigenetic priming, differentiation and gene regulation within the tissue architecture. These technologies are of great interest in life science and biomedical research.
© 2023. The Author(s).
Conflict of interest statement
R.F., D.Z. and Y.D. are inventors of a patent provisional disclosure related to this work. R.F. is scientific founder and advisor of IsoPlexis, Singleron Biotechnologies and AtlasXomics. The interests of R.F. were reviewed and managed by Yale University Provost’s Office in accordance with the university’s conflict of interest policies. The remaining authors declare no competing interests.
Figures
Comment in
-
Layering epigenomic and transcriptomic space.Nat Rev Genet. 2023 May;24(5):273. doi: 10.1038/s41576-023-00596-8. Nat Rev Genet. 2023. PMID: 36973496 No abstract available.
-
Harnessing the potential of spatial multiomics: a timely opportunity.Signal Transduct Target Ther. 2023 Jun 12;8(1):234. doi: 10.1038/s41392-023-01507-3. Signal Transduct Target Ther. 2023. PMID: 37302998 Free PMC article. No abstract available.
References
-
- Chen, A. et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell185, 1777–1792 (2022). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U54 AG079759/AG/NIA NIH HHS/United States
- U54 CA274509/CA/NCI NIH HHS/United States
- RF1 MH128876/MH/NIMH NIH HHS/United States
- UG3 CA257393/CA/NCI NIH HHS/United States
- U54 DK106857/DK/NIDDK NIH HHS/United States
- RF1MH128876/MH/NIMH NIH HHS/United States
- U54 AG076043/AG/NIA NIH HHS/United States
- U24 CA248453/CA/NCI NIH HHS/United States
- R01 CA245313/CA/NCI NIH HHS/United States
- UG3CA257393/CA/NCI NIH HHS/United States
- UH3 CA257393/CA/NCI NIH HHS/United States
- U54AG076043/AG/NIA NIH HHS/United States
- U54CA274509 /CA/NCI NIH HHS/United States
- R01CA245313/CA/NCI NIH HHS/United States
- U54AG079759/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
